
Obrázek 1: Konkrece se vyskytují na rozsáhlých plochách oceánského dna v hloubkách 3600-4400 metrů. V konkrecích se navíc vyskytuje dalších přibližně 50 prvků Mendělejevovy tabulky.
Chemické procesy se používají při zpracování chudobných nebo komplexních a těžko upravitelných rud, při zpracování rud obsahujících stopové prvky, ale i některých sekundárních surovin, odpadů, kde není vždy možné použít běžné úpravnické postupy na získání kovových koncentrátů.
Při zpracování sekundárních surovin se stále více uplatňují právě chemické způsoby úpravy. Recyklace druhotných surovin s obsahem kovů je podmíněna jednak nedostatkem primárních surovin, jednak nutností ochrany životního prostředí. Jedná se o celosvětový problém. Problematika zpětného získávání kovů z druhotných surovin a snižování ekologické zátěže cestou recyklace je proces značně složitý. Vyžaduje komplexní přístup a využívá většinou kombinace více metod a postupů při zpracování jednotlivých druhů primárních nebo sekundárních surovin.
Tato skripta by měla ukázat stručný přehled chemických, pyrometalurgických, hydrometalurgických a biotechnologických postupů a technologií používaných zejména při získávání kovů z rud a druhotných kovonosných surovin u nás i ve světě.
Nerostné suroviny budou i v 3. tisíciletí základní složkou materiální výroby. Jejich potřeba bude, na rozdíl od předchozích let, výrazně ovlivňována jak komplexem změn, které budou probíhat zejména v rozvojových zemích, tak taky větším důrazem na ochranu životního prostředí v souvislosti se stále více se prosazující zásadou trvale udržitelného rozvoje. Kvantitativní úroveň těžby a kvalita využívání nerostných i druhotných surovin je obecně závislá na stupni ekonomického a technologického rozvoje společnosti konkrétního státu. Státní správa vymezuje těžbu surovin pomocí zákonů a státních norem, popřípadě také norem oborových nebo podnikových. Jsou také stanoveny emisní limity do odpadních vod a ovzduší, tj. podmínky pro minimalizaci poškozování pracovního a životního prostředí a kontrolní mechanismy i sankce za neplnění stanovených podmínek.
Jednou z důležitých vlastností zásob nerostných surovin je jejich značně nerovnoměrné rozmístění ve světě. Např. 50 % prokázaných zásob zemního plynu důležitých pro zásobování Evropy je v Rusku na západní a severní Sibiři. Obdobně 50 % známých zásob ropy se nachází na ropných polích nacházejících se v arabských zemích na Středním východě, které představují rozlohou 5 % celkové rozlohy světa.
V České republice se v současnosti získávají ušlechtilé kovy pouze importem a zpracováním druhotných surovin. Zásoby některých nerostných surovin, vyskytujících se na našem území, byly do značné míry vyčerpány.
Ve společnosti Kovohutě Příbram a.s. se vyrábějí stříbrné anody jako vedlejší produkt výroby olova. Mezi další firmy zpracovávající druhotné suroviny s obsahem ušlechtilých kovů patří například Safina, a.s., Vestec, MHM eko, s.r.o., Zábřeh na Moravě, RECOM Brno, s.r.o. a další společnosti, které se zabývají výkupem dentálních kovů a slitin, elektronického odpadu, vyřazených autokatalyzátorů a podobně.
V současnosti ČR prakticky nemá využitelné zásoby rud a má omezené zásoby minerálních paliv. Má však dostatečné zásoby nerudních a stavebních surovin, jejichž životnost dosahuje řádově desítek až stovek roků (tab.1).
V poslední době je u nás také široce diskutována možnost využití primárních surovinových zdrojů zlata.
Surovinová základna je v České republice dnes tvořena několika geologicky studovanými lokalitami a to:
· Voltýřov - s předpokládanou povrchovou těžbou, zásoby rudniny jsou okolo 1 miliónu tun s obsahem zlata 2,5 – 3 g/tunu,
· Mokrsko (západ) - s uvažovaným vytěžením 15% celkových zásob podpovrchovou těžbou, zásoby jsou tvořeny 2 milióny tun rudniny s průměrným obsahem zlata 2,7 g/tunu,
· Roudný - zásoby 10 miliónů tun rudniny s obsahem zlata 2-3 g/tunu,
· Kašperské Hory - žilný typ ložiska s 200 000 t rudniny, obsah Au je v průměru 6 g/tunu.
Zájem o naše zlato projevila celá řada zahraničních těžebních a zpracovatelských společností.
Primární surovinové zdroje stříbra v České republice již patří minulosti. Největší průmyslový význam měly izomorfní příměsi stříbra v galenitu, popřípadě dalších sulfidech. Z Jáchymova a z Příbrami pocházejí nejkrásnější agregáty ryzího Ag o hmotnosti až 30 kg. Naše stříbrné nerostné bohatství bylo v minulosti postupně vyčerpáno a vzhledem k uzavření českých rudných dolů se roční spotřeba stříbra, která je cca 125 tun, kryje dovozem.
Primární suroviny pro výrobu ušlechtilých kovů se dnes, jak už bylo řečeno, v ČR netěží. Jediným zdrojem zůstávají druhotné suroviny. Spotřeba a struktura spotřeby zlata a stříbra v průmyslové oblasti dosahuje v průběhu posledních pěti let výraznějších změn. U elektronického průmyslu je předpokládán roční nárůst spotřeby zlata kolem 3 % a u fotografického průmyslu byl pozorován největší nárůst spotřeby, která se v tomto odvětví pomalu blíží 1/3 z celkové spotřeby stříbra. Dá se předpokládat, že právě u fotografického průmyslu se tato spotřeba zastaví nebo sníží z důvodu digitalizace. U platiny, paládia a rhodia je zaznamenán významný vzestup celkové spotřeby. U platiny představuje nárůst cca 4 %, u paládia 1,6 % a u rhodia téměř 5 % ročně.
V oblasti rudních surovin (železných i neželezných kovů) v dohledné budoucnosti prakticky neexistují perspektivy získávat surovinu z vlastních zdrojů a to z důvodů ekonomicky neefektivní těžby. Těžba chudých domácích zdrojů (Fe, Cu, Pb, Zn, Sn, W, Au, Ag), která byla možná jen s dotací, byla ukončena k 1.1.1994. Výhradní ložiska nerostných surovin ČR jsou uvedeny v tabulce 1.
|
Tabulka 1: Výhradní ložiska k 31.12.1998 - zásoby průmyslové a geologické |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Území ČR je z pohledu rud geologicky poměrně podrobně prozkoumáno. Do budoucna sice nelze vyloučit nález malých rudních ložisek lokálního významu, nicméně limitujícím faktorem pro jejich otvírku budou chybějící investičně náročné úpravárenské kapacity a střety se zájmy ochrany životního prostředí. Veškerá potřeba železných i neželezných rud v ČR je tedy kryta dovozem nebo se získává z druhotných surovin.
Potřeby kovů nebo jejich koncentrátů budou do budoucna záviset na strukturálních změnách v hutním a těžkém strojírenském průmyslu. Předpokladem pro výhodné získávání kovů v časovém horizontu 15-20 let je podpora aktivit vedoucích k využívání nerostných surovin z mořského dna. Pozornost byla především upřena na netradiční rudní zdroje - hlubokomořské polymetalické konkrece, železo-manganové kobaltem bohaté kůry a masivní sulfidické rudy, které mají vysoké obsahy neželezných kovů - manganu, mědi, niklu, zinku, ale i strategických kovů - kobaltu a molybdenu (obrázek 1). Výsledky průzkumu zúčastěných renomovaných institucí a ústavů byly velmi příznivé.
Přestože se polymetalické konkrece vyskytují na mnoha místech dna světových moří, rozsáhlé akumulace odpovídající jejich průmyslovému využití byly zjištěny pouze na několika lokalitách, především v oblasti Tichého oceánu. Největší plošnou ekonomicky využitelnou akumulací konkrecí na oceánském dně, v hloubkách 3800 - 4800 m, tvoří pás východně-západního směru, uvnitř zlomového pásma Clarion - Clipperton (CCZ) v subtropické části severního Pacifiku (obrázek 2). Celková rozloha zabírá 9 milionů km2 s odhadovaným množstvím zásob v miliardách tun této svým složením unikátní suroviny, která nemá ekvivalent na kontinentálních ložiscích. Hlavními kovy jsou především železo, mangan, měď, kobalt a nikl. Mezi doprovodné kovy pak patří zinek, molybden a skupina vzácných zemin - lanthanoidy.
|
|
|
Obrázek 1: Konkrece se vyskytují na rozsáhlých plochách oceánského dna v hloubkách 3600-4400 metrů. V konkrecích se navíc vyskytuje dalších přibližně 50 prvků Mendělejevovy tabulky.
|
|
|
|
Obrázek 2: Tektonická zóna Clarion-Clipperton [www.mpo.cz] (1 Japonsko, 2 Francie, 3 Rusko, 4 Čína, 5 Korea, 6 IOM, 7 ISA, 8 OMA, 9 OMI-1, 10 OMI - 2, 11 LMS) |
V této oblasti začaly hlubokomořský průzkum nově vzniklá účelová společenství - konsorcia (OMA - Ocean Mining Associates, OMI - Ocean Managment Inc., Lockheed Martin Systems Inc. a další), v nichž mají kapitálové podíly americké a západoevropské firmy a rovněž průkopničtí investoři, jejichž aktivity jsou dotovány ze státních rozpočtů a jsou zastoupeny vládními organizacemi. Mezi ně patří Japonsko (MMAJ); Francie (Genomod—Ifremer); Jižní Korea (KORDI); Čína (COMRA); Německo, Rusko a východoevropské státy (Česká republika, Slovensko, Polsko, Bulharsko, Rusko, ale také Kuba) sdružené do mezinárodní organizace Interoceanmetal (IOM). IOM bylo založeno v roce 1987 a bývalé Československo se stalo zakládajícím členem a zároveň jediným vnitrozemským státem na světě, který se aktivně podílel na hlubokomořském průzkumu nerostného bohatství oceánů. Sídlem IOM je polské přístavní město Štětín. Pro úplnost je třeba zmínit ještě dalšího průkopnického investora - Indii, která však směřuje své zájmy mimo hlavní oblast výzkumu hlubokomořských konkrecí. Německo v minulosti provádělo průzkum v Peruánském bazénu v jižním Pacifiku a v roce 2005 požádalo za náhradu o přidělení území v oblasti Clarion - Clipperton, zatímco Indie požádala o přidělení území v rámci další významné akumulace hlubokomořských konkrecí v centrální oblasti Indického oceánu. Základním právem ČR, jako podílníka IOM, je její přístup k nerostným surovinám na vyhrazeném průzkumném území na dně Tichého oceánu. Průzkumné území stanovil pro IOM Mezinárodní mořský úřad (ISA) v roce 1992. ISA řídí průzkumné a těžební aktivity, spojené s využíváním nerostných zdrojů na mořském dně. Jako člen ISA je ČR povinna přijmout vnitřní právní předpis respektující mezinárodněprávní zásady využívání nerostných zdrojů z mořského dna. Výše podílu jednotlivých členských zemí IOM vychází z jejich ročních vkladů, které pro ČR činí od roku založení IOM (1987) 136 mil.Kč. K tomu je nutno přiřadit i hodnotu „know-how“ získanou v průběhu expedic do oblasti svého průzkumného území. Tato hodnota dosud není určena. Stát po privatizaci svého podílu přestane poskytovat příspěvky na chod IOM, ponechá si však možnost usměrňovat aktivity soukromých podnikatelských subjektů v souladu s mezinárodním právem.
2. Stručný popis vybraných neželezných kovů
V této kapitole jsou charakterizovány některé chemicko-fyzikální vlastnosti základních kovů, metody jejich úpravy, využití a současný stav jejich produkce na území Evropské unie.
|
|
|
Obrázek 3: Chalkopyrit |
Rafinovaná měď se vyrábí z primárních surovin (chalkopyrit (FeCuS2), který je zachycen na obrázku 3, chalkozín, kovelin, bornit, tetraedrit, kuprit, malachit, azurit) a sekundárních surovin. Výrobkem rafinérií je měděná katoda. Ta se roztaví, leguje a dále zpracovává na tyče, profily, dráty, plechy, roury atd. Okolo 55 % vsázky, která se dodává do rafinerií mědi, je nakoupeno na mezinárodním trhu ve formě měděných koncentrátů, surové mědi nebo „šrotu“. Zbytek 45 % přichází z domácích měděných koncentrátů (odpady s obsahem mědi, elektroodpad).
Kovový odpad prochází nejprve předúpravou v kovošrotech nebo prostřednictvím hutí. Recyklace je na vysoké úrovni, protože měď se může vracet do operací beze ztráty svých původních vlastností a je k dispozici mnoho druhotných surovin. Měď je často legovaná zinkem, cínem, niklem, hliníkem a ostatními kovy za vytvoření široké palety mosazí a bronzů.
|
|
|
Obrázek 4: Bauxit |
Primární hliník se vyrábí z bauxitu, který je směsicí několika minerálů zahrnující hydroxidy hliníku, gibbsit, böhmit, diaspor, oxid hlinitý dihydrát (Al2O3·2 H2O) a oxidy železa (obrázek 4). 100 tun bauxitu poskytne 40-50 tun Al2O3, který potom dává 20-25 t Al. Většina bauxitu se těží mimo Evropu, ale v rámci Evropy existuje několik zařízení na výrobu oxidu hlinitého.
Průmysl druhotných surovin je závislý na zdrojích kovových odpadů. Ten může vznikat během výroby, továrního zpracování a odlévání výrobků nebo amortizací, která vzniká z vyřazených předmětů na konci doby jejich životnosti. Odpadní hliník je ze 100 % recyklován. Dodávka surovin do průmyslu EU je do značné míry naplňována domácí výrobou Al2O3 a recyklací kovových odpadů. Celkový výnos kovu však nedostačuje potřebám zpracovatelského průmyslu a v současnosti naplňuje pouhých 55 % požadavků EU. Výroba hliníku z druhotných surovin je v EU jednou z největších na světě.
|
|
|
Obrázek 5: Sfalerit |
Zinek se vyrábí z řady zinkových koncentrátů pyrometalurgickými a hydrometalurgickými pochody. Používají se kombinace pražení, loužení, elektrolýzy a destilace. Některé koncentráty obsahují vysoké podíly olova, které se také získává. Zinek se často přidružuje ke kadmiu a Zn-koncentráty jsou pak zdrojem i tohoto kovu. Zn-koncentráty nyní pokrývají méně než 25 % požadavků na rafinaci v EU a deficit se nahrazuje rostoucími dovozy. V současné době vzrůstá kapacita důlní těžby v Severní Americe, Austrálii a v některých zemích Jižní Ameriky. Na obrázku 5 je zachycen minerál Sfalerit (ZnS), který patří mezi primární zdroje zinku.
Sekundární (druhotné) suroviny, jako jsou odpady z galvanizace (pozinkování), popely, stěry, kaly, prach ze spalin oceláren a ze zpracování mosazi, jsou také zdrojem Zn. Výroba zinku v EU ze sekundárních zdrojů činí podstatný podíl z celkového množství výroby rafinovaného kovu. Recyklace Zn a výrobků s obsahem Zn je klíčovým problémem průmyslu.
|
|
|
Obrázek 6: Galenit |
Rafinované olovo pochází z primárního materiálu ve formě sulfidické olověné rudy galenit (PbS), koncentrátů nebo druhotných surovin. Primární výroba je založená na tavení rud s obsahem Pb za vzniku olověné slitiny, která se potom rafinuje. Ekonomika výroby primárního olova z rudy je vázána na obsahy stříbra a zinku v rudném ložisku. Většina hutí primárního olova má proto komplex rafinačních úprav pro rekuperaci stříbra a zinku obsažených v rudách. Obsah zinku a stříbra v rudách je hlavním ziskem zpracovatelů. Na obrázku 6 je zachycen sulfidický minerál galenit.
Poněvadž je v EU primárních zdrojů olova málo, dodává více než 50 % spotřeby olova průmysl druhotné rafinace. Olověné kyselinové akumulátory do automobilů jsou hlavním zdrojem (80 %) pro sekundární zpracování (Kovohutě Příbram, a.s.).
|
|
|
Obrázek 7: Greenockit |
Existuje pouze několik kadmiových hornin, jako greenockit (CdS) nebo otavit (CdCO3). Žádný z těchto minerálů však není průmyslově významný. Kadmium je ale téměř vždy obsaženo v rudách zinkových a někdy i v rudách olovnatých, ze kterých se rafinuje elektrolýzou. Minerály Zn, které obsahují Cd jako isomorfní složku, jsou již při koncentracích okolo 0,2 % pro získávání kadmia rentabilní. Kadmium se získává z pyrometalurgické rekuperace Pb-Cu (z prachu odplynů z tavících pochodů) nebo ze surového zinku. Na obrázku 7 je zachycen greenockit.
Prach z výstupního plynu se obvykle louží s H2SO4, aby se oddělilo Cd, které se potom vysráží jako CdCO3 nebo se vyredukuje jako kadmiová houba s více než 90 % Cd. Kadmiová houba se potom buď žíhá s NaOH, destiluje ve vakuu nebo se rozpouští a podrobuje elektrolýze, čímž se vyrobí Cd o vysoké čistotě (≥ 99,99 %).
Surový zinek se může destilovat v kolonách, aby se vyrobil čistý Zn a slitina Cd-Zn s více než 60 % Cd. Recyklují se hlavně baterie, aby se získalo Cd a Ni.
|
|
|
Obrázek 8: Zlato |
Doly ve všech částech světa odesílají velká množství ušlechtilých kovů v podobě surové rudy nebo ve formě vedlejších produktů do rafinerií EU. Evropská unie má největší rafinační a výrobní kapacitu pro ušlechtilé kovy na světě, i když jejich současné minerální zdroje jsou velmi omezené. Také recyklace ušlechtilých kovů z kovonosných odpadů byla v EU vždy důležitým zdrojem suroviny. Rafinerie ušlechtilých kovů s významnými kapacitami se nacházejí v Belgii, Německu, Švédsku, Finsku a Velké Británii. Ty obvykle rekuperují ušlechtilé kovy z rud olova, zinku, mědi nebo niklu, stejně jako z nízkojakostních kovonosných odpadních materiálů všeho druhu a dodávají čisté kovy v tyčích nebo deskách, zrnech nebo jako houbu.
V Evropě existují malá ložiska rud ušlechtilých kovů. Tyto zdroje činí okolo 4,5 % světového primárního stříbra, 1,1 % světového primárního zlata a 0,08 % primárních platinových kovů na světě. Ryzí zlato je zachyceno na obrázku 8.
V Evropě je mnoho společností, které se specializují na sběr, přepracování a obchod s kovonosným odpadem. Běžnými položkami jsou vyřazené tištěné spoje, zastaralé počitače, staré fotografické filmy, roentgenové desky a roztoky, vyčerpané elektro-pokovovací lázně atd.
Rafinace zlata, stříbra a skupiny platinových kovů se v EU provádí buď ve společnostech specializujících se na rafinaci a zpracování ušlechtilých kovů nebo v rafineriích základních kovů. Celková kapacita společností zabývajícími se v EU rafinací ušlechtilých kovů je největší na světě. Obchod s klenoty má nejvyšší spotřebu zlata a obchodování s fotografickým materiálem zase stříbra. Nejvyšší spotřeba platiny je při výrobě katalyzátorů do automobilů. Další hlavní využití jsou chemikálie, dentální zboží a investice, jako jsou například ražení mincí.
|
|
|
Obrázek 9: Cinnabarit (rumělka) |
Rtuť je jediným kovem, který se vyskytuje v kapalném stavu při pokojové teplotě a má ze všech kovů nejnižší body tání a varu. Objevuje se v přírodě ve formě cinnabaritu (rumělky-HgS), který se přidružuje k velmi těžké hlušině jako je kvarcit a čedič. Rtuť je také přítomna ve formě dalších sloučenin, jako jsou oxidy, sírany, chloridy nebo selenidy. Tyto sloučeniny jsou však poměrně vzácné a mají obecně malý význam. Existují výjimky, jako je livinstonit (HgSbS), který se využíval v Mexiku. Jakost primárních rud výrazně kolísá od 0,1 % až do více než 3 %. Na obrázku 9 je možno vidět cinnabarit (rumělku).
V případě bohatých rud (nad 2 % Hg), se za předúpravu považuje pouze drcení a prosévání a rozdrcená ruda může být vsazena přímo do pece. V případě chudých rud okolo 0,5 % Hg se používá flotace, aby se odstranila křemičitá hornina a získaly se koncentráty s průměrným obsahem rtuti 70 %. U hornin s nižším obsahem než 0,1 % Hg se ruda po rozdrcení praží. Rozklad rumělky se dosahuje při teplotě 600 oC. Kovová rtuť kondenzuje při pokojové teplotě. Oxidační pražení lze provádět buď v muflové či rotační peci.
Ostatní zdroje rtuti jsou rudy a koncentráty jiných kovů jako je měď, olovo, zinek atd. Rtuť se získává při čištění plynů, které jsou emitovány během výroby těchto kovů. Rtuť se také rekuperuje z druhotných surovin, jako jsou zubní amalgam, baterie apod.
|
|
|
Obrázek 10: Wolframit |
Název „těžkotavitelné kovy“ se vztahuje ke skupině kovů (v některých případech i kovů vzácných zemin), které se mohou charakterizovat převážně stejnými fyzikálními vlastnostmi. Těmito vlastnostmi jsou pro většinu těžkotavitelných kovů vysoký bod tavení, vysoká hustota, zvláštní elektrické vlastnosti, netečnost (inertnost) a zejména schopnost udělovat při malém přídavku do oceli a jiných kovů výjimečný nárůst fyzikálních schopností. Mezi tuto skupinu kovů patří chrom, mangan, wolfram, molybden, tantal, titan, niob, rhenium, hafnium a zirkonium. Typickým minerálem obsahujícím wolfram je wolframit ((Fe,Mn)WO4), který je zobrazený na obrázku 10.
Těžkotavitelné kovy se mohou vyrábět ze široké palety primárních a sekundárních surovin. Z primárních surovin se vyrábějí při hydrometalurgickém zpracování oxidických a sulfidických rud a koncentrátů, dále pak redukcí vodíkem a nauhličením, aby se získaly slinuté karbidy.
Výroba z druhotných surovin se zakládá na kovonosných odpadech s obsahem těžkých kovů a odpadech z výrobních pochodů, jako jsou vypotřebované katalyzátory apod. Recyklace hraje významnou úlohu a například 30 % světové dodávky wolframu je vyrobeno z druhotných surovin. Zpracovatelský průmysl wolframu je schopen zpracovat téměř každý druh kovonosného odpadu s obsahem wolframu, popřípadě jiných přidružených prvků.
|
|
|
Obrázek 11: Sylvín |
Alkalické kovy, které chemicky náleží do první skupiny periodické tabulky prvků, zahrnují kovy jako lithium, sodík, draslík, hořčík a rovněž extrémně vzácné radioaktivní francium. Alkalické kovy jsou charakteristické svým nízkým bodem tavení (tání) a hustotou. Mají stříbřitě bílou barvu a jsou měkčí než ostatní kovy. Alkalické kovy mají jen jeden pohyblivý elektron ve valenční sféře. V důsledku toho jsou vysoce reaktivní zejména s kyslíkem nebo vodou, kdy mohou reagovat bouřlivě za vzniku plynného vodíku a tepla.
Kovy alkalických zemin jsou podobné alkalickým kovům, ale reagují méně prudce s vodou. Jsou to prvky druhé skupiny periodické tabulky. Podle jejich rostoucího atomového čísla a metalurgické a technické důležitosti to jsou vápník a stroncium.
Typickým minerálem obsahujícím draslík je sylvín (KCl), který je zachycen na obrázku 11.
Kovové lithium se vyrábí velmi podobným způsobem jako kovový sodík. Výroba se provádí elektrolýzou roztavené eutektické směsi chloridu lithného (LiCl) a chloridu draselného (KCl) při teplotě okolo 450 oC. Jedním z představitelů minerálů, které obsahují lithium je fylosilikát lepidolit (KLi2AlSi4O10(OH,F)2).
Sodík jako kov a jeho sodné sloučeniny se využívají v širokém měřítku v průmyslu při výrobě chemikálií, ve farmacii, při hutních pochodech a rozličných dalších produktech každodenní potřeby. Kovový sodík se vyrábí běžně elektrolýzou taveniny chloridu sodného (NaCl). V přírodě se nachází například ve formě halitu (NaCl).
Draslík se vyskytuje v mnohých křemičitanových horninách. Hlavním komerčním zdrojem je naleziště soli (sylvín). Kovový draslík je stříbřitě-bílé barvy a byl prvním kovem, který byl izolován elektrolýzou. V průmyslovém měřítku je kovový draslík vyráběn pouze redukcí chloridu draselného (KCl) kovovým sodíkem.
Hořčík je stříbřitě bílý, kujný, chemicky reaktivní kov. Může se vyrábět elektrolýzou chloridu hořečnatého (MgCl2), což se provádí ze suroviny jako je dolomit, mořská voda, magnezit (CaCO3), karnalit, solné roztoky nebo z dolomitu, který se redukuje ferrosiliciem nebo hliníkem při tepelně-redukčním pochodu. Hořčík se také rekuperuje z druhotných surovin ze široké palety kovonosných odpadů s obsahem kovového hořčíku.
|
|
|
Obrázek 12: Pentlandit |
Nikl je prvek, který se v přírodě vyskytuje hlavně ve formě sulfidu, oxidu nebo jako prvek v křemičitých horninách. Získává se z minerálů lateritu ([(Fe,Ni)O(OH)]), pentlanditu (Fe,Ni)9S8, který je zachycen na obrázku 12, nebo pyrhotinu (Fe1-xS), kde se často vyskytuje jako příměs Ni (výjimečně až 7 %) ve formě izomorfní příměsi nebo mikroskopických inkluzí pentlanditu. Sulfidy niklu se často vyskytují společně s ekonomicky rekuperovatelným množstvím Cu, Co, Au, Ag, skupiny platinových kovů a několika dalšími kovy.
Niklové minerály vznikly také vlivem povětrnostních podmínek z ultrabazických hornin, které původně obsahovaly velké
množství Ni. Během času se nečistoty z ložisek vyplavily a nikl zůstal jako komplex oxidu křemíku, železa a hořčíku.
Minerály obsahující Ni se nacházejí v 25 zemích, včetně Kanady, Nové Kaledonie, Řecka, Austrálie, Ruska atd.
Metalurgie komplexů niklu se odráží v širokém pásmu extrakčních a rafinačních pochodů, které se provozují. Sulfidické rudy s obsahem Ni se mohou obvykle několikrát zkoncentrovávat relativně levnými úpravárenskými technikami předtím, než se koncentrát taví a rafinuje na produkty niklu.
Rudy lateritu (oxidická ruda) naopak podléhají pouze omezenému obohacování fyzikálními metodami, např. magnetickými technikami, nebo technikami gravitačního rozdružování a tudíž téměř původní objem rudy musí jít přímo do hutních závodů. Zpracování lateritu tak směřuje k vyšším nákladům, ale naopak náklady na těžbu jsou obvykle mnohem nižší, než u sulfidických rud.
Kobalt je oproti niklu zastoupen na Zemi i ve vesmíru ve výrazně menším množství. V přírodě nejsou známa naleziště rud s převažujícím množstvím kobaltu. Ten vždy pouze doprovází niklové rudy a nalezneme jej i jako doprovodný prvek v sulfidických rudách mědi nebo olova. Nejdůležitější nerosty kobaltu jsou smaltin (CoAs3), lineit ((Co, Ni)3S4) a kobaltin (CoAsS). Z oxidických kobaltových minerálů je nejrozšířenější asbolan ((Co,Mn)O.MnO2.4H2O.Fe2O3), kombinovaný hydratovaný oxid a heterogenit (Co2O3.H2O). V ryzím stavu je možné nalézt kobalt v množstí 0,5 - 2,5 % v železných meteoritech. Omezený výskyt kobaltu a složitý způsob výroby, který závisí na chemickém složení dané horniny, se pak odráží na vysoké ceně tohoto kovu. Největším zásoby rud s významným podílem kobaltu jsou v Rusku, Číně, Austrálii, Demokratické republice Kongo, Zambii a Zaire.
Nerostné suroviny, v našem případě rudy, se těží jen zřídka v takové formě, aby se mohly bezprostředně zpracovat. S užitným nerostem se vytěží i značné množství průvodních jalových nerostů a hornin, které je třeba od sebe navzájem oddělit. Některé druhy vytěžené nerostné suroviny jsou zase z hlediska kvalitativního složení dostatečně bohaté na užitkové minerály, avšak dalšímu zpracování nevyhovuje velikost zrna. Souhrn všech procesů, kdy je vytěžená rudná surovina převáděna do metalurgicky zpracovatelné formy nebo formy schopné odbytu se nazývá úprava rud nebo úpravnictví. Úpravou rudných surovin se získávají cenné složky v podobě vysokoprocentních koncentrátů vhodných pro následný odbyt. Jedním z úpravnických postupů zpracování rud je chemická úprava nebo-li chemické loužení. Úpravnické technologické postupy, za účelem získávání kovů, mohou být vztaženy i na druhotné suroviny, jako jsou kovonosné odpady, elektroodpad apod.
Pro chemickou úpravu suroviny je charakteristické dlouhotrvající působení chemického činidla až do úplného rozpuštění a vyloužení užitkové složky - kovu, při čemž základní odpadní (jalová) látka suroviny zůstává nezměněná. V některých případech je cílem vyloužit nečistoty z koncentrátu a zvýšit tak koncentraci užitkové složky - kvalitu koncentrátu.
K tomu, aby mohly být rudy chemicky zpracovávány, musí být většinou v předúpraveném stavu. Předúprava zahrnuje otevření rudy – drcení a mletí a někdy i předběžnou chemicko-fyzikální úpravu spojenou s odstraněním specifických nečistot (viz. Přípravné procesy), kam patří:
a) pražení rudy
b) specifická chemická úprava, kterou se převedou užitkové složky do rozpustné formy nebo pomocí které se zabrání velké spotřebě loužicího roztoku.
K základním procesům chemických metod úpravy (add b) patří:
a) loužení, při kterém se užitkové složky rudy selektivně rozpouštějí vhodným činidlem,
b) dělení výluhu od tuhého zbytku odpadu, a to usazováním, zahušťováním, filtrací, promýváním a čířením,
c) příprava výluhu na srážení užitkové složky (kovu) selektivní extrakcí nebo iontoměniči
d) srážení užitkové složky (kovu) z roztoku cementací, elektrolýzou, krystalizací, hydrolýzou a jinými metodami,
e) zpracování sraženiny filtrací, sušením,
f) recirkulace loužícího roztoku po jeho regeneraci do stavu vhodného na opětovné použití pro loužení.
Volba vhodného přípravného postupu si vyžaduje dokonalé informace o chemickém a mineralogickém složení rudy, znalost složení základních minerálů rudy a způsob jejich vzájemné vazby. Podmínkou loužení užitkové složky je alespoň její částečné obnažení, které se zabezpečí přiměřeným drcením a mletím. V některých případech je před chemickým zpracováním rudy potřebná její předkoncentrace některou z běžných úpravnických metod rozdružování.
Pražení je přípravný pyrometalurgický proces před hydrometalurgickým zpracováním. Metoda přímého loužení je možná jen při některých oxidech, uhličitanech, síranech nebo ryzích kovech. Už jednoduché sulfidy se těžko louží. Špatně se louží také křemičitany a chemicky stálé oxidy.
Do lehce loužitelné formy se minerály převádějí většinou vhodným pražícím postupem, který má za úkol převést těžko loužitelné sloučeniny na sloučeniny rozpustné ve vodě nebo v jiném rozpouštědle, případně přeměnit nežádoucí složky na nerozpustné látky a zabránit tak znečištění výluhu. Pražení se také používá k aglomeraci (spojování) částic koncentrátu po flotaci do agregátů vhodných ke zpracování například v šachtových pecích.
Z hlediska probíhajících chemických reakcí lze pražicí postupy rozdělit do několika skupin, a to na termický rozklad, oxidační pražení, sulfatační pražení, chloridační pražení a redukční pražení.
· Termický rozklad (kalcinace) - od ostatních pražících postupů se odlišuje tím, že pro průběh reakce není potřebná plynná fáze.
Příkladem je rozklad uhličitanů:
MeCO3 → MeO + CO2
Kde Me je ion kovu.
Například smithsonit (ZnCO3) se před loužením kalcinuje, protože vzniklý ZnO je v procese loužení reaktivnější.
V některých případech nemusí termický rozklad způsobit změnu chemického složení vsázky, jeho účelem může být pouze narušení krystalické mřížky minerálu s případným následujícím prudkým ochlazením – dekrepitace (např. ZnO2).
· Oxidační pražení - pro chemickou úpravu je to mimořádně významný proces. Tímto postupem se sulfidy a kovy přemění za přístupu vzduchu na oxidy. Při oxidačním pražení se síra S2- nahrazuje kyslíkem O2-. Vznikají nové sloučeniny rozpustné ve vodě nebo v kyselinách. Teplota pražení je 600 °C a více. Oxidační pražení se používá před loužením v kyselém prostředí. Průběh oxidace sulfidů charakterizují rovnice:
MeS + 3/2 O2 → MeO + SO2 (plyny se z pece odsávají)
nebo
MeS + 2 O2 → MeSO4 … MeSO4 → MeO + SO3
reakce oxidace sulfidů jsou exotermické, to znamená, že se při těchto reakcích uvolňuje teplo a může se dosáhnout zápalné teploty, při které pak proces probíhá samovolně. Jako příklad je možno uvést oxidaci pyritu nebo arzenopyritu.
4 FeS2 + 11 O2 → 2 Fe2O3 + 8 SO2
2 FeAsS + 5 O2 → Fe2O3 + As2O3 + 2 SO2
Oxidační reakce je závislá na vysoké teplotě (okolo 700°C), kdy se pak udržuje v chodu vlastním uvolněným teplem. Uvolněný oxid siřičitý (SO2) se používá k výrobě kyseliny sírové (H2SO4).
· Sulfatační pražení je pražení, při kterém se sulfidy přeměňují na daleko rozpustnější sírany. Je třeba dbát na to, aby proběhla oxidační reakce:
MeS + 2 O2 → MeSO4
Nesmí však nastat následný rozklad vzniklého síranu, proto se teplota pražení udržuje na teplotě nižší než 600 °C. Pro počáteční fázi oxidace má podstatný význam zápalná teplota sulfidu kovu. Zápalné teploty některých sulfidů jsou uvedeny v tabulce 2:
|
Tabulka 2: Zápalné teploty některých minerálů |
||||||||||||||||||||||||
|
||||||||||||||||||||||||
Jako příklad sulfatačního pražení můžeme uvést získávání Ni z pentlandit-pyrhotinové rudy, pražení chalkopyritu (CuFeS2) při teplotě 650 °C a převedení sulfidové sloučeniny mědi na rozpustnou síranovou formu
2 CuFeS2 + 7,5 O2 → 2 CuSO4 + Fe2O3 + 2 SO2
nebo selektivní sulfatační pražení komplexních sulfidických rud obsahujících pyrit, sfalerit, galenit a sulfidy mědi na rozpustné sírany ZnSO4 a CuSO4.
·
Chloridační pražení
spočívá na reakci mezi chloridy alkalických kovů nebo kovů alkalických zemin a
oxidy nebo sulfidy kovů při zvýšené teplotě
v oxidační atmosféře. Využívá se tak vysoká aktivita chlóru vůči kovům,
jejich oxidům nebo jiným sloučeninám, v důsledku kterých vznikají chloridy.
Principiální reakce:
Me2+ + 2 NaCl → MeCl2 + 2 Na+
· Redukční pražení je typické před zpracováním oxidických rud s obsahem Ni a Co, popřípadě dalších kovů, hydrometalurgickým postupem - loužením v roztoku NH3+ + CO2. Tento typ pražení může být vhodný i ke zpracování hlubokomořských konkrecí, které mají podobné složení. Cílem pražení je selektivní redukce oxidů žádaných neželezných kovů, přičemž železo (nebo mangan v konkrecích) zůstává v oxidické formě. Surovina je redukována buď uhlíkem, nebo technicky použitelnými plynnými redukovadly. Účelem redukčního pražení je tedy převedení oxidů kovů na nižší oxidy nebo na čistý kov. Redukce na nižší oxid má význam jen tehdy, když je tento oxid lépe rozpustný v loužícím roztoku. Jako příklad je možno uvést redukci kassiteritu.
SnO2 + 2 C → Sn + 2 CO
Redukční pražení je také možno využít při výrobě olova:
2 PbS + 3 O2 → 2 PbO + 2 SO2 (oxidační pražení)
PbO + CO → Pb + CO2 (redukční pražení)
V této kapitole jsou popsány jen vybrané druhy pražících pecí, které se používají při úpravě rud před dalším zpracováním.
Šachtové pece v běžném vyhotovení se v rámci přípravy chemické úpravy rud málo využívají, protože vyžadují hrubozrnou vsázku. Na chloridační pražení se používají upravené šachtové pece použitelné na pražení peletizovaných koncentrátů.
Rotační pece se v chemické úpravě také málo využívají, a to při relativně hrubozrné vsázce. Jejich použití je omezené pro některé postupy redukčního pražení, při kterých vsázka částečně slinula, dále tam, kde je třeba pracovat při teplotách nad 1000 °C a pro spékací procesy. Hlavními příklady je výroba oxidu zinečnatého ve Waelzově peci (typ rotační pece), výroba výpražku pro výrobu feroniklu, kalcinace hydroxidu hlinitého a kalcinace hydroxidu hořečnatého.
Jinými aplikacemi jsou předúpravy rozličných surovin při vysokých teplotách, spalování fotografického filmu a foto-papíru nebo vysoušení koncentrátů a směsí materiálu při nízkých teplotách. Rotační pec je znázorněna na obrázku 13.
|
|
|
Obrázek 13: Rotační pec |
Etážové pece mají tvar svislého válce, který je uvnitř předělený vyzděnými klenbami na více etáží, kterých počet je podle velikosti pece a potřebného času pražení 4 až 14. Ve velkých pecích je celková užitečná plocha etáží až 500 m2. Schéma etážové pece je na obrázku 14. Hrabla jsou natočená tak, že střídavě shrnují pražený materiál k obvodovým nebo centrálním otvorům etáží, kterými materiál přepadne na nižší etáž. Tak vsázka postupuje od nejvyšší etáže směrem dolů dle šipek. Teplota jednotlivých pater pece je regulovatelná a na určitých hodnotách se udržuje vhodným uspořádáním průtoku horkých plynů a hořáky umístěnými na každé etáži pece.
|
|
1 – válcové těleso 2 – klenba 3 – otáčející ramena 4 – hrabla 5 – otáčející se hřídel 6 – výpusť
|
|
Obrázek 14: Etážová pec |
Fluidní pece jsou určené pro pražení ve vznose a používají se jen pro tepelné samonosné exotermické reakce - prakticky jen na oxidační pražení. Příkladem pece tohoto druhu je Nicolsova-Freemanova pražící pec (obrázek 15).
Pece na pražení ve vířivé (fluidní) vrstvě (obrázek 16) jsou charakteristické tím, že částice umístěné na pórovité desce (obrázek 17), kterou protéká proud plynu, získávají při určitých rychlostech plynu specifické fyzikální vlastnosti. Vrstva prachových částeček podstatně zvětší svůj objem a chová se v určitém smyslu jako proudící kapalina. Tento jev zanikne, klesne-li rychlost proudění plynu pod určitou hranici rychlosti označovanou jako fluidizační práh. Pokud rychlost proudění plynu překročí určitou maximální hranici fluidizace, plyn strhává částečky materiálu a vynáší je ven z reaktoru. Při fluidizačním stavu jsou částice hmoty v dokonalém styku s použitým nosným plynem.
|
|
1 - sušící etáž 2 - hrablové shrnovače 3 -
kanál přívodu koncentrátu 4 - zapalovací zařízení 5 - hořáky
6 - přívod sekundárního vzduchu 7 - odvod spalin
|
|
Obrázek 15: Nicolsova-Freemanova pec na pražení ve vznosu |
|
|
1 – šroubovicový dopravník 2 – přívod vzduchu 3 – rošt 4 – boční otvor směřující do chladícího zařízení 5 – odvod spalin
|
|
Obrázek 16: Fluidní pec |
|
|
|
Obrázek 17: Schéma roštu ve fluidní peci |
Rošt fluidní pece je děrovaná plošina, vrchní část vzduchových trysek přechází do kuželovitých výpustí, do kterých zapadají žáruvzdorné kuličky fungující jako spalný uzávěr trysek. Nevýhodou těchto pecí je potřeba předsušovat koncentráty před jejich vsázením do pece.
Tuto nevýhodu odstranila konstrukce fluidních pecí Dorr, do kterých se vstřikuje materiál ve formě rmutu s obsahem 50 % tuhých částic. Výhodou těchto pecí je, že koncentráty není třeba filtrovat a do pece se vstřikují přímo ze zahušťovačů.
Fluidní pece jsou zvláště vhodné, když se vyžaduje dobrá regulace teploty například pro sulfatační pražení a dokonalé pražení Zn koncentrátů, stejně jako při kalcinaci oxidu hlinitého. Při pražení se dosahuje teplot 900-1000 ˚C. Pro laboratorní účely slouží k pražení muflové pece (obrázek 18).
|
|
|
Obrázek 18: Muflová pec LAC při teplotě 1000 ˚C |
Loužení je selektívní získávání jedné nebo více složek z tuhého substrátu do kapalného výluhu. Z fyzikálně-chemického hlediska je to heterogenní proces, kterého se zůčastňují nejméně dvě fáze: tuhá (s) a kapalná (l).
Volba způsobu a podmínek převodu užitkové složky z tuhé do kapalné fáze předpokládá poznání základních informací o dynamice a kinetice procesu. Při rozpouštění tuhé látky se rozpadá tuhé skupenství materiálu za vzniku homogenní kapalné směsi s nižším stupněm uspořádanosti. Aby se rozpouštěla tuhá látka v kapalném rozpouštědle, je třeba narušit síly, které udržují částice v tuhém skupenství. Při rozpouštění iontových krystalů ve vodě je třeba dodat energii na přerušení kohézních sil tuhé fáze na uvolnění jednotlivých iontů ze struktury. Tato energie se rovná vazebné energii krystalové mřížky a její teoretickou hodnotu je možné určit výpočtem. Interakce částic rozpouštěné látky s molekulami rozpouštědla se nazývá solvatace, pokud je rozpouštědlem voda, jedná se o hydrataci.
Rozpustnost látky v daném rozpouštědle je jednoznačně určená teplotou, tlakem a koncentrací jiných látek přítomných v roztoku. Rozpustnost tedy můžeme ovlivňovat změnou teploty nebo přídavkem dalších složek. Pokud je rozpouštění tuhých látek v kapalinách endotermický děj, zvyšování teploty má jednoznačně příznivý vliv na rozpustnost. Proto se některé těžko rozpustné látky musí loužit za zvýšené teploty.
Užitková složka se do roztoku může převést:
a) rozpadem organizovaného tuhého skupenství rozpouštěné látky účinkem rozpouštědla – při tomto procese se chemická podstata složek roztoku nemění,
b) substituční chemickou reakcí základních stavebních částic tuhé fáze s ionty roztoku kyselin, zásad nebo solí za vzniku nové sloučeniny rozpustné ve vodě.
Rychlost rozpouštění lehce rozpustných minerálů a jiných tuhých látek se většinou neurčuje rychlostí průběhu chemické reakce na hranici tuhá fáze – kapalina, nýbrž rychlostí difuzních procesů. Na povrchu se vytvoří vrstvička, jejíž koncentrace se rovná koncentraci nasyceného roztoku. Přes tuto vrstvičku probíhá difůze rozpustných látek. Rychlost průběhu difůze určuje rychlost celého procesu. Transportní procesy v kapalinách a plynech jsou difůze a konvekce.
Difůze je založená na tepelném molekulárním pohybu. Konvekce spočívá na makroskopických pohybech jako jsou turbulence a laminární proudění. V obou případech musí existovat koncentrační spád, aby nastala látková výměna. Difůze je možná jen v neturbulentním prostředí. Přechod rozpouštědla z roztoku k minerálnímu povrchu, jako i přechod produktů reakce do celé masy roztoku, probíhá cestou konvekce a na hranici s minerálem probíhá účinkem tepelné molekulární difůze. Během rozpouštění tuhé látky ubývá při jejím povrchu rozpouštědlo, které se spotřebuje na rozpuštění vyloužené složky ve vrstvě, která přiléhá k částicím látky, ale také působením na jiné přítomné částice zpracovávané rudy. Matematická závislost (experimentálně ověřená) byla objevena v polovině minulého století. Vychází z těchto veličin (obrázek 19):
Množství látky dQ (rozpouštědlo) difundující za jednotku času dt válcem s příčným průřezem S při gradientu koncentrace dc/dx, přičemž gradient dc/dx znamená, že v příčném řezu se souřadnicí x se koncentrace rovná c a v příčném řezu x+dx se rovná c+dc. Fick vyjádřil celý průběh rovnicí:
dQ= -D.S dc/dx*dt
|
|
|
Obrázek 19: Schéma procesu difůze |
V rovnici pojmenované Fickův zákon difůze je součinitel difůze D a jeho hodnota je vyjádřena:
D=RT/N * 1/3 pmd
R – plynová konstanta (8,314)
T – absolutní teplota (K)
N – Avogádrovo číslo (6,02 . 1023)
m - viskozita rozpouštědla (Pa.s)
d – průměr částic difundované látky (m)
Znaménko mínus ve Fickově rovnici znamená, že difůze proběhla ve směru snížení koncentrace difundující látky, tj. ve směru záporného gradientu koncentrace.
Difůze v hydrometalurgickém loužení probíhá v povrchové vrstvě roztoku na rozhraní minerálu a roztoku (obrázek 20). Okolo povrchu rozpouštějícího se nerostu se nachází vrstva roztoku (d = 20 – 50 mm) se sníženou koncentrací látek ubývajících při procesu rozpouštění. Velikost této vrstvy se mění v závislosti na relativní rychlosti pohybu minerálu a roztoku. Hloubka difuzní vrstvičky d závisí v podstatné míře na fyzikálních podmínkách rozpouštění a hlavně na podmínkách promíchávání rmutu. Při loužení jemně pomletých rud má rychlost promíchávání menší význam.
|
|
|
Obrázek 20: Zjednodušený proces difuze a konvekce při loužení |
Pro charakteristiku činitelů, které určují podmínky loužení, je vždy důležité vědět, jestli rychlost dané reakce je určená difuzí nebo rychlostí vzájemného chemického působení činidla a minerálu. Difůze obvykle probíhá pomaleji, a protože kontrolním činitelem rozpouštění je nejpomalejší děj, reakce probíhá obvykle v difůzní oblasti. Rychlost rozpouštění je dána změnou koncentrace rozpouštěné látky uvnitř roztoku za časový úsek dc/dt. Maximální rychlost rozpouštění je na začátku procesu, při nasycenosti roztoku je rychlost rozpouštění nulová.
S nárůstem teploty se zvyšuje rychlost chemické reakce i rychlost difůze. To vyplývá z toho, že s růstem teploty obsahuje většina částic rozpouštědla dostatečné množství energie, a proto snadněji rozrušují nebo oslabují chemické vazby rozpustných látek.
Rychlost difůze se zvětšuje přibližně o 2% při zvýšení teploty o 1%. Vliv teploty na rychlost loužení se hodnotí na základě hodnoty tepelného součinitele, kterým se rozumí zvýšení rychlosti reakce rozpouštění při nárůstu teploty o 10oC. V případě heterogenních reakcí řízených procesy difůze je tepelný součinitel reakcí menší nebo se rovná 1,5. Proces loužení se může urychlit i zvyšováním tlaku za určitých podmínek. Při rozpouštění minerálů představujících chemické sloučeniny s iontovou vazbou krystalické mřížky není převládajícím činitelem rychlosti rozpouštění rychlost difůze, nýbrž kinetika chemické reakce ve vzájemném působení s činidlem. Příkladem je rozpouštění CuO v kyselině sírové.
Z činitelů, kteří určují rychlost rozpouštění rudných nerostů v hydrometalurgických procesech, mají největší význam:
1. koncentrace rozpouštědla
2. koncentrace oxidačního činidla (pokud je oxidace potřebná k rozpouštění minerálu)
3. teplota
4. podmínky míchání nebo dodávání čerstvého roztoku
5. poměr mezi krystalickou a koloidní částí rozemleté rudy
6. vztah množství roztoku k množství zpracovávané rudy
7. hmotnost rudy
8. přítomnost hydrofobních látek v roztoku, které se adsorbují na povrchu rudy
9. škodlivé příměsi v roztoku
Obzvlášť významné jsou vlastnosti minerálů, hlavně charakteristika jejich krystalické mřížky. Velmi důležitou charakteristickou podmínkou rozpouštění je poměr hmotnosti roztoku spotřebovaného při loužení k hmotnosti zpracované rudy. Tento poměr má vliv na rychlost rozpouštění a dobu promíchávání. Rychlost rozpouštění se během loužení mění. Proto mluvíme buď o rychlosti rozpouštění v daném okamžiku v=f(t), kde t je čas nebo o rychlosti průměrné (střední) vprum během některého časového úseku rozpouštění dt. Množství kovu M, vylouženého za dobu t je úměrné rychlosti rozpouštění.
Vzájemné působení činidel s minerály je dáno složením a strukturou krystalové mřížky minerálů. Ve struktuře mřížky známe několik vazeb:
1) iontová vazba – je to nejjednodušší a zároveň nejvýznamnější chemická vazba atomů minerálů. Dochází k ní tak, že 1 atom ve snaze zaujmout stabilní konfiguraci nejbližšího inertního prvku plynu odevzdá přebytečné elektrony z valenčního obalu jinému atomu, který si tak doplní nestabilní atomovou sféru na stabilní opět nejbližšího inertního prvku plynu. Změnou elektrického náboje prvku se mění jeho poloměr (velikost), kationty a anionty na sebe navzájem působí elektrostatickou přitažlivou silou F podle Coulombova zákona.
F= 1/ξ . (e1 . e2)/(r1 + r2)2
ξ konstanta prostředí
e1, e2 náboje kationtu a aniontu
r1, r2 poloměry kationtu a aniontu
Minerály s iontovou vazbou jsou lehce rozpustné, při substituční reakci vytváří kationt se sloučeninou – sloučeninu rozpustnou ve vodě, jako například:
CuO + H2SO4 → CuSO4 + H2O
2) kovalentní vazba – vzniká mezi atomy s nepárovými elektrony a antiparalelně orientovanými spiny. Tento typ vazby je charakteristický pro stejný druh atomů.
3) kovová vazba – spočívá v tom, že atomy pro svou vazbu používají své vlastní elektrony. Každý atom, který se účastní této vazby odevzdá elektrony, stává se tak kationtem a vazba vznikne mezi kationty a volně se pohybujícími elektrony. Vazební síly působí rovnoměrně na všechny nejbližší prvky.
Při rozpouštění kovové látky je nutné převedení kovu do iontového stavu (například elektrolýzou).
Ag0 → Ag+ + e-
4) molekulární vazba – tu vytvářejí sloučeniny vzájemně orientovaných molekul v krystalických strukturách. Jsou to vazby nejslabší. Krystaly, které mají mezi sebou tyto vazby, mají nízký bod tání, malou tvrdost, vysoký koeficient tepelné roztažnosti a velmi malou nebo žádnou vodivost (například grafit).
5) smíšené vazby – je to spojení několika druhů vazeb v jednom minerálu. Tyto sloučeniny jsou tvořeny například klastry atomů či sítěmi polyedrů vázaných silnými kovalentními vazbami a navzájem spojenými slabšími vazbami iontovými či van der Waalsovými. Takové struktury se nazývají heterodesmické (na rozdíl od látek homodesmických tvořených pouze jedním druhem vazeb). Minerály se smíšenými vazbami jsou například sulfidy, oxidy, selenidy, teluridy, arzenidy a jiné. Mezi těmito minerály existují vazby iontové, kovalentní a kovové.
Vliv viskozity rmutu na rozpouštění
Viskozita je fyzikální veličina, udávající poměr mezi tečným napětím a změnou rychlosti v závislosti na vzdálenosti mezi sousedními vrstvami při proudění skutečné kapaliny. Zvětšením viskozity (míry vnitřního tření) kapaliny se snižuje rychlost difůze iontů molekul. Největší vliv na difůzi má viskozita jemně dispergovaných rmutů. Větší částice vznášející se ve rmutu nemají vliv na jeho viskozitu a nesnižují tak rychlost rozpouštění.
Vliv oxidace
Pro procesy, u kterých je průběh podmíněný oxidací, má význam rozpustnost kyslíku v tekuté části rmutu. V hustějších rmutech probíhá zvýšení koncentrace kyslíku rychleji, než ve zředěných rmutech, ale ne v takové míře, jako v základním rozpouštědle. To se vysvětluje tím, že při promíchávání vstupuje do roztoku kyslík z nasávaného nebo profukovaného vzduchu.
Při působení kyslíku na povrch kovů, jejich slitin a sulfidů probíhají tyto děje:
· Fyzikální adsorpce plynů,
· Aktivní adsorpce s fixací kyslíku na povrchu,
· Tvoření kyslíkového filmu,
· Silná oxidace povrchu za vzniku oxidického povlaku.
Loužení sulfidů s kyslíkem jako oxidačním činidlem se využívá při získávání Cu, Zn, Ni, Co a kyselých nebo zásaditých roztoků ze sulfidických koncentrátů nebo hutnických kamínků. Za nepřítomnosti kyslíku jsou sulfidy (železných i neželezných kovů) do 300°C ve vodě nerozpustné, ale za přítomnosti kyslíku přecházejí do roztoků. V závislosti na teplotě a pH roztoku probíhá reakce loužení s tvorbou elementární síry nebo iontů SO42-.
Závislost rozpouštění na geometrickém tvaru, zrnitosti a stupni otevření zrn louženého materiálu
Volba postupu hydrometalurgického zpracování rudy se určuje především možností nejdokonalejšího vytěžení cenných složek a závisí na formě, ve které se cenná složka nachází, na její asociaci s jinými složkami rudného materiálu a na fyzikální stavbě rudy. Tvar zrn nerostu určuje rychlost rozpouštění, čímž se stává jedním z činitelů podmiňujícím efektivnost loužení a objem zařízení na loužení. Podle rozličných tvarů zrn je možno vybrat několik typických geometrických tvarů charakterizujících buď celé zrno nebo jednotlivé elementy jeho povrchu. Můžeme si představit pět základních tvarů znázorněných na obrázku 21 (a, b1, b2, c a d):
|
|
|
Obrázek 21: Typy prorůstání zrn a kinetika jejich rozpouštění |
a) Zrno má plochý tvar, přičemž jeho tloušťka je nepatrná v porovnání s délkou a šířkou. V tomto případě bude závislost množství Q přecházejícího do roztoku od doby t graficky velmi blízká přímce.
b) Částice nerostu - zrno je vrostlé do horniny a rozpouštění probíhá jen z jedné strany. Proto tento případ jsou možné dvě varianty:
b1) Stěny dutiny, ve kterých je zrno uzavřené, jsou téměř paralelní. Povrch rozpouštění je téměř stálý. V tom případě se rychlost rozpouštění skoro nemění; závislost rozpouštění na čase je graficky vyjadřená křivkou blízkou přímce. Úhel jejího úklonu je menší než v případě a).
b2) Stěny dutiny uzavírající minerál jsou takové, že obvod rozpouštějícího se vrostlého zrna není stejný. V tomto případe se křivka, která charakterizuje rozpouštění, odkloňuje ve větší nebo menší míře od přímky.
c) Zrna mají kulovitý povrch. V takovém případě se rozpustí za stejnou dobu kulová vrstva stejné tloušťky, ale každá další vrstva patří kouli menšího průměru a obsahuje teda menší množství látky než předcházející vrstva. Křivka, která charakterizuje rozpouštění kulovitých zrn, na začátku strmě stoupá, ale potom se stoupání postupně zmírňuje. Pokud jsou částice nerostů kuličky rozdílného průměru, ukončí se rozpouštění jemnějších zrn dříve než zrn hrubších.
d) Zrna vrostlá do horniny jsou nepravidelná, odkrytá jsou jen na malé plošce. Příslušná křivka rozpouštění bude přiměřeně měnit tvar. Z toho vyplývá, že na rozdíl od fyzikálních metod úpravy rud není potřebné při hydrometalurgickém zpracování úplné uvolnění rozpouštěného minerálu od mechanického spojení s jinými minerály tvořícími rudu. Při loužení postačí jen částečné odkrytí, které umožňuje rozpouštět užitkový minerál.
Stupeň odkrytí zrn minerálu se měří poměrem hmotnosti volných částeček minerálu k celkovému množství v rudě. Tento poměr se vyjadřuje v procentech.
Optimální zrnitost pomleté rudy, která zabezpečuje nejvýhodnější stupeň odkrytí, se zjišťuje experimentálně na základě pokusů loužení rudy při různé zrnitosti. Pro zjišťování optimální zrnitosti rudy se určují tito činitelé:
výtěžnost kovu z rudy,
spotřeba činidel,
náklady na dodatečné pomletí, aby měla ruda požadovanou zrnitost.
Velmi užitečným doplněním k pokusům tohoto druhu je mikroskopický výzkum rudy a produktů úpravy.
Defekty v krystalech zvyšují chemickou aktivitu tuhých látek. Tato skutečnost se využívá při intenzifikaci procesu loužení. Aktivace je možná mechanickým nebo termickým způsobem (pražením).
Při drcení a mletí se zvyšuje specifický (měrný) povrch pevných látek, deformace a částečné narušení krystalické mřížky, které způsobuje vysokou koncentraci dislokací a atomových defektů.
Výzkum kinetiky mletí poukazuje na tři etapy změn rozměrů částeček a specifického povrchu mleté látky v závislosti na čase (obrázek 22).
|
|
|
Obrázek 22: Závislost změny zrnitosti r a specifického povrchu S od času mletí t |
Ze začátku se rozměry částic rychle zmenšují a současně narůstá specifický povrch. Po dosáhnutí určitého stupně disperzity začíná agregace (shlukování) částic, což způsobí, že se rozměry částic začnou zvětšovat až do ustálení určitého rovnovážného stavu. Agregaci částic způsobují van der Waalsovy síly.
Přírůstek volné entalpie – Gibbsové energie se zvětšuje v důsledku šířící se destrukce krystalické mřížky až do úplného přechodu do látky amorfního charakteru.
Pro průběh reakcí mechanicko-chemické aktivace tuhé látky jsou možné dvě varianty:
1. Tuhá látka se aktivuje jemným mletím, po kterém následuje reakce s rozpouštědlem (loužícím činidlem),
2. Chemická reakce probíhá současně s mechanickou aktivací (mletím, roztíráním)
Druhý případ je energeticky výhodnější. Kromě toho se zabraňuje tvorbě případných povlaků v tuhé fázi, které by brzdily proces reakce, poněvadž se povrch částic stále obnovuje.
a) jednoduché rozpouštění – uplatňuje se při krystalických minerálech s iontovou vazbou nebo při některých minerálech s velmi deformovanou krystalickou mřížkou. Působení polárních molekul vody současně s tepelnou energií iontů v mřížce zabezpečí rozrušení mřížky a zvětšení vzdálenosti mezi ionty.
Příklady reakcí:
MeCl(s) + voda → MeCl (l)
MeSO4 (s) + voda → MeSO4 (l)
b) výměnná reakce – výměnná reakce oxidů kovů nebo solí s kyselinou, zásadou nebo roztoky solí.
MeCO3(s) + H2SO4 → MeSO4(l)+ H2O + CO2
CuO(s) + H2SO4 → CuSO4(l)+ H2O
3 CuO(s) + 2 FeCl3 + 3H2O → 3 CuCI2(l)+ 2 FeOH3
Fe2O3(s) + 6 HCl → 2 FeCl3(l)+ 3 H2O
c) oxidace kationtu nebo aniontu – chemické vazby v mřížce se narušují změnou elekronického složení atomu, rozměrem atomu a tím i změnou sily a charakteru reakce mezi atomy. Výsledkem je vytvoření nové sloučeniny. Někdy se rozpouštění minerálu oxidačně – redukční reakcí uskuteční oxidací kationtu kovu do stavu vyšší valence. Například při uranu:
U3O8 + 4 H2SO4 + MnO2 (oxidační činidlo) → 3 UO2SO4(l) + MnSO4 + 4 H2O
3 U3O8 + 9 H2SO4 + NaCIO3 (oxidační činidlo) → 9 UO2SO4(l) + NaCI + 9H2O
Sloučenina uranu U3O8, což je podvojný oxid uranično-uranový (U2O5.UO3) se tak mění na síran uranylu (UO2SO4) – vysoce rozpustnou formu.
Další příklad rozpouštění spojeného s oxidací je loužení zlata (tato reakce je zároveň i komplexotvorná):
4 Au(0) + 8 NaCN + 2 H2O + 02 → 4 NaAu(I)(CN)2(l) + 4 NaOH
d) rozpouštění s vytvářením komplexních sloučenin – například amoniakální loužení kovové mědi.
2 Cu+O2+n(NH3) → 2 CuO . n(NH3)(l)
2 Cu+2 CuO.n NH3 → 2 Cu2O . n(NH3) (l)
Cu + 2 NH4OH + (NH4)2CO3 → Cu(NH3)4CO3(l)+ 3 H2O
Cu + Cu(NH3)4CO3 → Cu2(NH3)4CO3(l)
Proces loužení založený na tvorbě rozpustných komplexů je charakterizovaný vysokou selektivností. Nejčastěji používanými loužicími roztoky jsou voda, H2SO4, HCI, NaOH, NH3, Na2CO3, (NH4)2CO3, NaCN a KCN. Pokud je jalovinou (odpadem) uhličitan, není vhodné použít k loužení kyselinu.
Mnohé minerály, jako halit (kamenná sůl), sylvín (draselná sůl), karnalit, modrá skalice a další se při normální teplotě rychle rozpouštějí ve vodě.
V roztocích kyseliny sírové se rozpouští kovová měď, oxidy mědi a zinku a celestin (SrSO4).
V roztocích kyseliny chlorovodíkové se rozpouští sfalerit (ZnS), molybdenit (MoS2) a dioptas (CuSiO2(OH)2).
Některé minerály se rozpouštějí ve čpavku, jako například wolframit [(Fe,Mn)(WO4)] a erytrin [Co3(AsO4)2 · 8H2O].
V roztocích alkálií se rozpouští bauxit (směs hydroxidů hliníku) a zinkit (ZnO).
V roztocích sulfidu sodného (Na2S) a chloridu železnatého (FeCl2) se rozpouští cinabarit (HgS), antimonit (Sb2S3) a pyrargyrit (Ag3SbS3). Sulfidy těžkých neželezných kovů mohou být louženy vodným roztokem síranu železitého [Fe2(SO4)3]. (Tabulka 3)
|
Tabulka 3: Podmínky loužení některých minerálů |
|||||||||||||||||||||||||||
|
Tato kapitola popisuje jednotlivé typy loužení a jejich parametry. Patří sem technologie loužení průsakem, technologie loužení s promícháváním, tlakové a bakteriální loužení.
Tento způsob se používá při zpracování rud, které se dají lehce loužit při hrubém mletí umožňujícím průsak roztoku vrstvou rudy. Podle zrnitosti zpracovávaného materiálu se rozlišují dva případy loužení průsakem:
1) loužení kusového materiálu získaného většinou dvoustupňovým nebo třístupňovým drcením (například rudy Cu),
2) loužení hrubých písků získaných vytříděním ze rmutu po rozemletí (například zlatonosné rudy).
Účinnost chemického loužení ovlivňuje řada činitelů jako jsou:
Průsaková (perkolační) rychlost
Je to rychlost průsaku loužicího média vyjádřena snížením úrovně hladiny roztoku nad pískovou náplní v nádobě za jednotku času. Měří se v centimetrech nebo milimetrech za hodinu. Závisí na:
1) pórovitosti materiálu,
2) stupni jílovitosti,
3) výšce hladiny v nádrži (tíha kapaliny).
Rychlost průsaku 3 cm · h-1, při loužení drobného materiálu, se považuje za dostatečnou, lepší je však začínat s rychlostí 8 cm · h-1 a více. Na podmínky průsaku rudy loužícím roztokem má podstatný vliv struktura jednotlivých kusů rudy (trhliny, póry, kapiláry).
Pro pórovitost e platí vztah:
e = (1- r0/r) . 100 %
e = r0 . (1/r0-1/r) . 100 %
kde e je pórovitost (%), r - hustota (kg/m3) a r0 - objemová hmotnost (kg/m3).
Například, když hustota rudy r=2,7 g/cm3, objemová hmotnost jejího písku r0= 1,35 g/cm3, potom pórovitost vypočteme ze vztahu:
e = 1,35(1/1,35 – 1/2,7)100 = 50%
Podstatný význam pro charakteristiku pórovitosti má mokrý nebo suchý způsob plnění perkolátorů, ale i uložení částeček, které jsou buď volné nebo stlačené.
Kapilární jevy
Vnikání rozpouštědla do pórů a kapilár v rudě je spojené s pohybem kapaliny v těchto průchodech a s difuzí samotného rozpouštědla a produktů vzájemného působení činidla s nerostem. Důležitou úlohu při loužení zaujímají kapilární jevy založené na smáčivosti minerálu rozpouštědlem. Rychlost pronikání horninou závisí od vytěsnění plynů z jejího povrchu, což závisí na smáčivosti horniny roztokem a rozpustnosti plynů v kapalině. Prosakování rozpouštědla horninou tedy závisí na hydrofobnosti jejího povrchu. Vliv hydrofilních látek se projevuje kladně při různých loužicích procesech, obzvláště při loužení hrubozrných materiálů. Všeobecně se rychlost průsaku zmenšuje se zmenšujícími se částicemi materiálu. Výška náplně rudy má také vliv na rychlost průsaku. Při zvýšení vrstvy rudy v nádrži nastává přiměřený pokles rychlosti průsaku.
Zářízení nádrží na loužení průsakem
Loužení prosakováním se provádí v dřevěných nebo ocelových nádržích válcového nebo pravoúhlého tvaru s plochými dny. Ocelové nádrže větších rozměrů se vyrábějí hranolové. Obsah nádrží je od 25 – 800 tun rudy. Na zhotovení dřevěných nádrží se používají dobře vysušené desky z borového nebo jedlového dřeva spojené ocelovými obručemi. Ocelové nádrže se zhotovují z hrubých ocelových plátů navzájem spojenými sváry nebo nýty a jsou trvanlivější než nádrže dřevěné. Prosakování roztoků v kádích se zabezpečuje dvojitými, tj. filtračními dny (obrázek 23). Někdy se na zrychlení průsaku zavádí odsávání roztoku mezi dnem kádě a filtrem.
|
|
|
Obrázek 23: Filtrační dno v perkolační nádrži 1 - dno, 2 - první řada trámů, 3 - druhá řada trámů, 4 - těsnící vložky, 5 - ochranná vrstva, 6 - vypouštěcí otvory, 7 - filtrační vrstva |
Loužení rmutu promícháváním (agitace) je v porovnání s loužením průsakem účinnější proces. Používá se pro loužení jemnozrných rud. Louží se v nádržích se zařízením na promíchávání (agitační nádrže). Jedním ze základních činitelů určujících náklady na loužení rmutu je specifická spotřeba energie. Jsou známy dva způsoby loužení:
nepřetržité – rmut postupuje řadou kádí zapojených za sebou. V těchto kádích dochází k intenzivnímu promíchávání a rmut se přečerpává z jedné kádě do druhé.
přerušované – používá se přerušovaného čerpání do souběžně pracujících kádí. Po zpracování se rmut přečerpává do sběrné kádě a loužicí kádě se plní novými roztoky.
Pokud nemůžeme zabezpečit tok rmutu samospádem, používají se na přečerpávání čerpadla.
K výhodám nepřetržitého provozu patří:
možnost automatizace činnosti,
menší spotřeba pracovníků,
menší průřez potrubí, menší mohutnost čerpadel a motorů na vyčerpávání rmutu,
· výhodnější využití loužicích nádrží.
Zařízení nádrží na loužení promíchávání
Promíchávání a zároveň oxidace rmutu rozpuštěným vzdušným kyslíkem se při loužení dosahuje mechanickým mícháním nebo pneumaticky. Podle toho se kádě s míchadly dělí (analogicky s flotací) na:
1. mechanické,
2. pneumatické,
3. pneumomechanické (kombinované),
Příkladem loužící nádrže s mechanickým promícháváním rmutu je nádrž s vrtulovou míchačkou (obrázek 24).
|
|
|
Obrázek 24: Příklad nádrže s mechanickým promícháváním s vrtulovou míchačkou |
V kádi jsou radiálně umístěné přepážky, které brání otáčení rmutu, způsobují vytvoření středové nasávací nálevky a obvodového převýšení rmutu, který se převaluje do střední části kádě. Rmut dopadá na dno kádě a proudí po stranách k povrchu. Taková cirkulace vzduchu za vzniku nasávací nálevky způsobuje dobrou aeraci v důsledku intenzivního promíchávání se vzduchem.
Konstrukce kádě se sací míchačkou nasávající rmut trubkou vznikla na základě využití principu míchadlové „kontaktní“ nádrže používané na promíchávání rmutu s flotačními činidly (obrázek 25). Štít nad míchadlem nejenže chrání míchadlo před usazováním materiálu při přerušení míchání, ale zmenšuje také tření a zamezuje tvoření velkých vírů, čímž se snižuje spotřeba energie.
|
|
|
Obrázek 25: Příklad nádrže s mechanickým promícháváním se sací míchačkou |
Ve vysokých cylindrických ocelových nebo dřevěných nádržích se rmut promíchává systémem mamutího čerpadla – aeroliftem. Nádrž má kuželovité dno. Během plnění se do nádrže vhání obvodovými trubkami vzduch. Tyto trubky sahají až ke dnu nádrže, aby se zabránilo sedimentaci rmutu. Poměr výšky kádě k průměru je 5/3. Kuželovité dno svírá s vodorovnou rovinou 60°.
Rozšířené jsou také nádrže s pneumaticko-mechanickým (kombinovaným) promícháváním. Příkladem nádrží tohoto typu jsou cylindrické loužicí nádrže s vrtulovým míchadlem a okrajovým aeroliftem (obrázek 26). Rmut se mechanicky promíchává vrtulovým míchadlem, jehož hřídel prochází středovou vratnou trubkou. Po obvodu vratné trubky jsou otvory pro cirkulaci rozvířeného rmutu. Část objemu rmutu cirkuluje pomocí čtyřramenného aeroliftu, který čerpá rmut ze dna v blízkosti stěny nádrže.
|
|
|
Obrázek 26: Loužící nádrž s pneumaticko-mechanickým promícháváním 1 - míchadlo, 2 - aerolift |
Uzavřené loužící nádrže na loužení pod tlakem (autoklávy) se používají na loužení při zvýšeném tlaku a teplotě, která převyšuje teplotu varu při atmosférickém tlaku (například loužení wolframových koncentrátů, výroba hliníku – Bayerova metoda, tlakové loužení sulfidických rud obsahujících zlato při 180°C apod.).
Autoklávy na loužení pod tlakem při současném zahřívání můžou být s povrchovým zahříváním a mechanickým promícháváním nebo můžou být s vnitřním zahříváním suchou parou, kterou se rmut i současně promíchává.
Autoklávy s povrchovým zahříváním a mechanickým promícháváním jsou horizontální, autoklávy s mícháním suchou parou jsou vertikální.
Vertikální autokláv (obrázek 27) má v zaoblené vrchní části otvor pro plnění rmutu. Po naplnění autoklávu po určitou výšku se horní plnící otvor uzavře a ze spod se přivádí suchá pára, která prochází rmutem, ohřívá ho a zároveň promíchává. Tlak suché páry je ze začátku 1,2 MPa a postupně se zvýší až na 1,5 MPa. Po uplynutí času potřebného pro loužení se uzavře i spodní otvor a otevře se spodní ventil na vnějším vyústění středové trubky, kterou se vyprazdňuje autokláv působením tlakového polštáře páry nad hladinou rmutu. Autoklávy pracují periodicky podle cyklu:
Plnění – ohřívání – vzrůst tlaku a loužení – vyprazdňování
Pro plynulou činost jsou potřebné alespoň tři cyklicky pracující autoklávy.
|
|
|
Obrázek 27: Vertikální autokláv 1 – rmut, 2 - suchá para |
Při těžebním způsobu dobývání rud vznikají ztráty – část zásob v rozsahu 10 – 50 % zůstává v zemi. Je to ruda zanechaná v ochranných pásmech, pilířích apod. Úplnému vydobytí brání technické a ekonomické podmínky. Dodatečné získání rud ze zbylých nevydobytých částí ložiska "in situ" aniž by se musely těžit na povrch umožňuje podzemní loužení (obrázek 28).
Podzemní loužení je možné jen při určitých příznivých geologických podmínkách. Záleží na charakteru rudy a doprovodných hornin, na úložných poměrech, na struktuře a textuře rudného tělesa. Vhodné je především pro sedimentační ložiska, ve kterých je přirozený součinitel filtrace dost velký na pronikání loužícího roztoku horninou. Pokud je hornina nepropustná, musí se uvolnit trhacími pracemi s velkou silou výbuchu.
Když dojdeme podle geologického průzkumu k závěru, že rudy je tolik, že se loužení vyplatí, tak očekáváme zvýšení výtěžnosti užitkové složky, snížení počtu pracovníků a snížení rizikovosti práce.
Postup podzemního loužení rud:
1) ložisko se rozloží na kvádry
2) voda prosakující z povrchu lomu se přivádí chodbou do sedimentační nádrže, kde se usazují nečistoty.
3) Z nádrže na povrch je vertikální vzdálenost 10 m, čímž se získává potřebný tlak pro kropení vodou,
4) Skrápěcí systém se skládá z perforovaných trubek a hlavního potrubí,
5) Loužící roztok proniká základkovým materiálem do chodeb, odkud se roztok s vysokým obsahem kovu přečerpává na horní patro a z něho dále na povrch,
6) Po určité době dochází k ucpání loužících pórů, a proto se musí plytkými rýhami obnovit jejich propustnost. Tato metoda se týká loužení zbytkového ložiska.
|
|
|
Obrázek 28: Průřez loužící haldou na Kounradském Cu-ložisku (Kazachstán) 1 - infiltrační rýhy, 2 - přívod roztoku H2SO4, 3 - vrty pro kyselinu, 4 - sběrný kanál výluhu, 5 – nádrž výluhu |
Na povrchu haldy jsou infiltrační rýhy vyhloubené buldozérem do hloubky 0,5 až 0,7 m. Sběrný kanál je hluboký 1 až 1,5 m. Louží se roztokem kyseliny sírové o koncentraci 5 g .l-1. Intenzita přívodu loužícího roztoku je 40 l.t-l, kovnatost výluhů je do 2,5 g .l-1 Cu. Potrubí jsou vyrobena z polyetylénu.
Metody podzemního hydrochemického dobývání užitkových nerostů, při kterých odpadá potřeba investičně nákladných hornických zařízení a zpracování velkých množství balastních látek, je známá již dlouhou řadu let. Při prvních pokusech těžby uranu podzemním loužením byl použit jako loužící médium roztok uhličitanu a hydrogenuhličitanu sodného (Na2CO3, NaHCO3). Tato metoda je však vhodná pouze pro ideálně uložené rudy, kde je uran vázán především v šestimocné formě a rudy jsou tak dobře loužitelné. Další možností je podzemní loužení uranu roztokem 5%-ní kyseliny sírové. Tento způsob však není vhodný, pokud ložisko vykazuje velkou reakční spotřebu kyseliny. Nedostatky předchozích způsobů byly řešeny použitím roztoků uhličitanů alkalických kovů s přídavkem oxidačního a komplexotvorného činidla. Protože charakter uranových ložisek se případ od případu značně liší, byly i pro metody podzemního loužení navrženy některé další postupy. V aplikacích provozního měřítka se však využívá prakticky výhradně základní varianty, pro kterou je charakteristické vtláčení a čerpání loužícího roztoku 5%-ní kyseliny sírové pomocí vtlačných a čerpacích vrtů (obrázek 29).
|
|
|
Obrázek 29: Schéma podzemního loužení uranu |
V České republice se již uran podzemním loužením nedobývá. Vytěžená uranová ruda je chemicky upravována na povrchu.
Bakteriální loužení spočívá v biokatalytickém urychlování oxidačních procesů v důsledku čehož se těžce rozpustné sulfidy oxidují na snadno rozpustné sírany. Mikroorganismy, které se používají pro bakteriální loužení můžeme rozdělit na:
a) Autotrofní (chemolitotrofní) organismy – energii získávají chemickou přeměnou anorganických látek (např. NH3, S, H2S, H2, sloučeniny Fe atd.). Zdrojem uhlíku, potřebného pro jejich růst, je CO2 získávaný ze vzduchu. Tyto bakterie jsou převážně mezofilní, tzn., že jsou aktivní při teplotě 30-35°C. Při teplotě nad 50°C bílkoviny obsažené v bakteriích koagulují, enzymy jsou neaktivní a buňky hynou. Jsou schopné přeměňovat dostupné anorganické sloučeniny na látky organické. Jsou také schopny fotosyntézou vázat světelnou energii v chemickou (zelené rostliny, řasy..).
K autotrofním bakteriím patří například zástupci rodu Acidithiobacillus.
b) Heterotrofní organismy – pro svůj růst využívají organické látky jako jsou organické kyseliny, uhlovodíky, proteiny apod. K těmto zástupcům patří například parazitické mikroorganismy, plísně a kvasinky.
c) Mixotrofní organismy - typ výživy některých rostlin, které jsou nebo mohou být současně nebo střídavě autotrofní (tj. fotosyntetizují) i heterotrofní (tj. přijímají organické látky z prostředí), např. masožravé rostliny, zelení bičíkovci, některé řasy.
Využití bakterií jako aktivátorů při rozpouštění minerálů nazýváme bakteriální nebo mikrobiologické loužení. Podle Gajdardzijeva spočívá bakteriální loužení na biokatalytickém urychlování oxidačních procesů, v důsledku čehož se oxidované sulfidy, které jsou mimořádně těžko rozpustné v roztocích kyselin, stávají ve formě síranů rychle rozpustnými.
Enzymy jsou většinou vysokomolekulární bílkoviny. Vyznačují se vysokou specifičností, přičemž katalyzují tvorbu nebo rozrušování některých druhů chemických vazeb. Katalytické působení enzymů je dáno aktivními centry molekul. Tato centra tvoří s molekulami substrátu aktivní komplexy. Jejich rozpadem vzniká finální produkt. Působení enzymů urychluje některé reakce 109 – 1014-krát.
Charakteristika bakterií druhu Acidithiobacillus ferrooxidans
Mikroorganismus Thiobacillus ferrooxidans byl původně izolován a popsán jednak jako samostatný rod Ferrooxidans a jednak jako příslušník rodu Acidithiobacillus. Ve všech případech byla bakterie izolována z kyselých důlních vod z nejrůznějších lokalit a to vždy v případech, když byla v minerálech přítomna síra nebo železo. První vyizoloval síruoxidující baktérie Beijerich v roce 1904.
Thiobacillus thiooxidans a Thiobacillus ferrooxidans, které izolovali Waxmann a Joff v roce 1922, se staly základními mikroorganismy v procesu loužení sulfidických rud (Borovec, 1989).
Buňky Acidithiobacillus ferrooxidans jsou drobné tyčinky (0,5 x 1 - 1,5 µm) se zaoblenými konci, nesporulující, gram negativní. Bakterie v mladé kultuře jsou pohyblivé, mají jeden polární bičík, který stará kultura ztrácí. Všechny druhy Acidithiobacillus jsou schopné využívat jako energetický substrát síru a její redukované anorganické sloučeniny. Finálním produktem oxidace těchto sloučenin je síra, některé druhy akumulují síru nebo polythionáty. Acidithiobacillus ferrooxidans je také schopen získávat energii oxidací Fe2+ na Fe3+. Jako zdroj C využívají tyto bakterie pouze atmosférický CO2. Stavba buňky této bakterie je popsána na obrázku 30.
V tabulce 4 můžeme vidět rozdělení thionových bakterií podle fyziologických vlastností.
|
Tabulka 4: Rozdělení thionových bakterií podle fyziologických vlastností (podle Kokala 1969) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
+ - bakterie substrát utilizující - - bakterie substrát neutilizující NT - nestanovováno |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Acidithiobacillus ferrooxidans získává energii k udržení vnitřní organizace a pro udržení životních pochodů oxidací podle rovnice:
Fe2+ → Fe3+ + e- + 11,3 kcal.mol-1
Je-li Fe2+ v roztoku v podobě FeSO4:
4 FeSO4 + 2 H2SO4 + O2 → 2 Fe2(SO4)3 + 2 H2O
Acidithiobacillus ferrooxidans využíva i Fe vázané v sulfidech, resp. i Fe elementární, které rozpouštějí. Oxidace Fe v roztoku probíhá za přítomnosti Acidithiobacillus ferrooxidans 1000x rychleji než stejná chemická reakce.
Oxidace síry: 2 S0 + 3 O2 + 2 H2O → 2 SO42- + 4 H+
Oxidace thiosíranů: 2 S2O32- → S4O62- + 2 e-
nebo S2O32- + H2O + O2 → 2 SO42- + 2 H+
Oxidace sulfidů kovů: 2 MeS2 + 2 H2O + 7 O2 → 2 MeSO4 + 2 H2SO4
Jako zdroj uhlíku Acidithiobacillus ferrooxidans využívá co2 z atmosféry. Energii k jeho asimilaci získává výše uvedenými oxidačními reakcemi. Tato energie je přenášena do míst fixace CO2 v buňce pomocí energetického přenašeče - adenozintrifofsfátu - ATP.
V případě Acidithiobacillus ferrooxidans probíhají tedy v buňkách 2 procesy:
produkce energie oxidací anorganických látek
fixace CO2 a následná tvorba organické hmoty.
|
|
|
Obrázek 30: Stavba těla bakterie Acidithiobacillus ferrooxidans |
Na povrchu buňky se nachází buněčná stěna, která slouží jako mechanická ochrana před nepříznivými vnějšími vlivy. Zodpovídá za tvar buňky, udržuje v buňce potřebnou vlhkost a vyrovnává osmotický tlak mezi buňkou a okolím. Je pevná, ale přitom i elastická a propustná pro soli, nízkomolekulární a některé vysokomolekulární látky. Buněčná stěna je pokryta slizovou vrstvou.
Vnitřní část buňky tvoří cytoplazmatický roztok obsahující jednotlivé orgány. Cytoplazma je koncentrovaný vodní roztok mnoha různých biomolekul. Vyplňuje vnitřní prostor buňky. Obsahuje 400 částic o průměru 20 nm (ribozómy) centra syntézy bílkovin.
Důležitým orgánem buňky je nukleoid nebo-li jádro. To obsahuje fibrily, které jsou tvořeny vlákny kyseliny deoxyribonukleové (DNA). Jádro nemá vlastní membránu, přesto je viditelně odděleno od cytoplazmy. Jádro má nepravidelný tvar a je nositelem genetických informací.
Dalšími nepostradatelnými orgány uvnitř těla buňky jsou ribozomy. Jsou to tělíska složená z RNA a bílkovin. Hlavní jejich úloha spočívá v syntéze různých typů bílkovin.
Cytoplazma buňky je ohraničena cytoplazmatickou membránou, která je uložena pod buněčnou stěnou. Má dvě důležité funkce:
reguluje propustnost buňky
účastní se v energetické přeměně, v její dýchací soustavě, přenosu elektronů.
V cytoplazmě se také vyskytují mesozomy a někdy také vnitrochromozomové molekuly DNA prstencového tvaru - takzvané plazmidy. Mesozomy pravděpodobně také ovlivňují počátek a průběh dělení buňky. Plazmidy jsou pravděpobně zodpovědné za adaptační schopnosti buňky. Neúčast plazmidů neovlivňuje životní procesy.
Mechanismus oxidace mikroorganismy
Mechanismus oxidace sulfidů mikroorganismy lze rozdělit na přímou a nepřímou cestu.
Přímá cesta
Přímé loužení je založeno na degradaci minerálů enzymatickou oxidací. Ve vodě nerozpustný minerál je oxidován na rozpustný v několika enzymaticky katalyzovaných mezistupních. K uskutečnění tohoto procesu je nutný přímý kontakt mezi povrchem minerálu a baktériemi. Podmínkou je, aby byl povrch minerálu před oxidací ve vodním prostředí disociován a takto uvolněný sulfidový anion mohl vstoupit do metabolismu bakterie:
MeS → Me2+ + S2-
Enzymatické systémy baktérií zachytí sulfidový iont a oxidují jej na síran.
S2- + 2O2 → SO42-
Tímto se posune rovnováha disociace ve směru rozpustného síranu, což umožňuje další disociaci nerozpustného sulfidu. Podmínkou reakce je přísun kyslíku.
MeS + 2 O2 → MeSO4
Oxidace pyritu: 2 FeS2 + 7 O2 + 2 H2O → 2 H2SO4 + 2 FeSO4
4 FeSO4 + O2 + 2 H2SO4 → 2 Fe2(SO4)3 + 2 H2O
Oxidace pyrhotinu:
4 FeS + 9 O2 + 4 H+ → 4 Fe3+ + 4 SO42- + 2 H2O
Oxidace chalkozinu:
2 Cu2S + O2 + 2 H2SO4 → 2 CuSO4 + 2 CuS + 2 H2O
Oxidace covellinu:
CuS + 2 O → CuSO4
Oxidace chalkopyritu:
4 CuFeS2 + 17 O2 + 2 H2SO4 → 4CuSO4 + 2 Fe2(SO4)3 + H2O
Oxidace arzenopyritu:
2 AsFeS + 7 O2 + 2 H2O → 2 FeAsO4 + 2 H2SO4
Oxidace molybdenitu:
2 MoS2 + 9 O2 + 6 H2O → 2 H2MoO4 + 4 H2SO4
Acidithiobacillus ferrooxidans může enzymaticky oxidovat i nesulfidické minerály, např. minerály uranu:
2 U4+ + O2 + 4 H+ → 2 U6+ + 2 H2O
2 UO2 + O2 + 4 H+ → 2 UO22+ + 2 H2O
2 U6+ + 4 H2O → 2 UO22+ + 8 H+
Oxidace U4+ katalyzovaná Acidithiobacillus ferrooxidans není hlavní reakcí při loužení uranových rud, protože v systému je vždy Fe3+ vznikající oxidací Fe2+ (jde zde více o nepřímé loužení).
Nepřímá cesta
K nepřímé extrakci kovů z rudy dochází při interakci minerálu s meziprodukty nebo koncovými produkty metabolismu mikroorganismů. Podstatou nepřímého loužení je oxidace substrátu síranem železitým za vzniku síranu extrahovaného kovu, síranu železnatého a u sulfidů taky elementární síry.
Bakterie Acidithiobacillus ferrooxidans opakovaně oxidují dvojmocné železo na trojmocné, které pak působí jako oxidační činidlo. Tyto přeměny probíhají čistě chemicky při aerobních i anaerobních podmínkách dle následujících rovnic:
MeS + 2 Fe3+ + H2 + 2 O2 → Me2+ + 2 Fe2+ + SO42- + 2 H+
Pyrit.:
FeS2 +Fe2(SO4)3 → 3 FeSO4 + 2 S0
Chalkopyrit:
CuFeS2 + 2 Fe2(SO4)3 → CuSO4 + 5 FeSO4+ 2 S0
Covellin:
CuS + Fe2(SO4)3 → CuSO4 + 2 FeSO4+ S0
Sfalerit:
ZnS + Fe2(SO4)3 → ZnSO4 + 2 FeSO4+ S0
Uraninit:
UO2 + Fe2(SO4)3 → UO2SO4 + 2 FeSO4
Malachit:
CuCO3.Cu(OH)2 + 2 Fe2(SO4)3 → 6 CuSO4 + 2 Fe2O3 + 3 CO2 + 3 H2O
Zdrojem Fe2(SO4)3 bývá nejčastěji pyrit. Ten se oxiduje ve dvou stupních:
1) 2 FeS2 + 7 O2 + 2 H2O → 2 FeSO4 + 2 H2SO4
2) 4 FeSO4 + O2 + 2 H2SO4 → 2 Fe2(SO4)3 + 2 H2O
Nejlépe dochází k oxidaci Fe2+ na Fe3+ v prostředí kyseliny sírové (pH = 1,5 - 5,0). Při oxidaci Fe2+ na Fe3+ pomocí Acidithiobacillus ferrooxidans se H2SO4 spotřebovává (viz výše popsaná rovnice)
Zvyšováním pH extrakčního roztoku vznikají různé hydrokomplexy železa:
Fe3+ + H2O → FeOH2+ + H+
FeOH2+ + H2O → Fe(OH)2+ + H+
Fe(OH)2+ + H2O → Fe(OH)30 + H+
a dále vznikají amorfní sraženiny neurčitého složení, které potom přecházejí na hydratované oxidy Fe3+ např. goethit--FeOOH. V prostředí kyseliny sírové vzniká při pH = 1,6 pufrovaný systém, vhodný k tvorbě nerozpustných komplexů se síranovým aniontem např. FeSO4+ ; FeOHSO40; FeHSO42+; případně nerozpustné produkty jarozitového typu:
3 Fe3+ + K+ + 2 HSO- + 6 H2O → KFe3(SO4)2(OH)6 + 8 H+
jarozit
Monovalentní K v jarozitu může být podle složení loužícího roztoku nahrazen Na+, NH4+.
Sraženiny trojmocného železa zabraňují kontaktu loužícího roztoku s povrchem minerálu, omezují aktivitu bakterií a při loužení hald nebo in-situ snižují propustnost horniny. Tvorbě sraženin je možno zabránit udržováním nízkého pH loužícího roztoku a nízké koncentrace Fe3+ v roztoku. Při loužení v nádržích nebo tancích se jarozit používá jako nosič baktérií. Bakterie na něm rostou rychleji než v roztoku. Na tomto principu pracuje BACFOX (BACterial Film Oxidation) v Jihoafrické republice.
Elementární síra se oxiduje baktériemi Acidithiobacillus thiooxidans nebo Acidithiobacillus ferrooxidans podle následující rovnice:
2 S0 + 3 O2 + 2 H2O → 2 H2SO4
Vzniklá H2SO4 rozpouští oxidické minerály a současně udržuje kyselé prostředí, vhodné pro růst bakterií.
Azurit:
2 CuCO3.Cu(OH)4+ 3 H2SO4 → 3 CuSO4 + 2 CO2 + 4 H2O
Chrysokol:
CuSiO3.2H2O + H2SO4 → CuSO4 + SiO2 + 3 H2O
Tenorit:
CuO + H2SO4 → CuSO4 + H2O
V praxi dochází k přímému i nepřímému loužení současně a záleží jen na druhu minerálu, který z mechanismů bude převažovat.
Galvanické rozpouštění
V podstatě jde o nepřímé loužení. Při fyzikálním kontaktu dvou odlišných sulfidických minerálů v elektrolytu se vytvoří galvanický článek. Elektrický proud prochází z minerálu s vyšším klidovým potenciálem k minerálu s nižším potenciálem.
Ze sulfidů má v prostředí H2SO4 nejvyšší potenciál pyrit (0,63 V vůči standardní H-elektrodě v 1 M H2SO4 ) a je v tomto článku katodou (zápornou elektrodou). Chalkopyrit (0,52 V) nebo sfalerit (-0,24 V) mají nižší potenciály a při styku s pyritem jsou anodou (kladnou elektrodou). Při galvanickém rozpouštění je povrch anody (chalkopyrit) disociovaný, přičemž do roztoku přecházejí uvolněné ionty z anody (Cu+, Fe+) a vzniká elementární síra:
CuFeS2 → Cu2+ + Fe2+ + 2 S0 + 4 e--
Po přenosu elektronu z anody na katodu a reakci vzdušného O2 s vodíkem z disociované H2SO4 vzniká na povrchu katody (pyritu) voda:
4 H+ + O2 + 4 e- → 2 H2O
Celý proces popisuje obrázek 31.
|
|
|
Obrázek 31: Schematické znázornění galvanického rozpouštění chalkopyritu v kontaktu s pyritem |
Galvanické rozpouštění chalkopyritu je možno vyjádřit sumárně:
CuFeS2 + O2 + 4 H+ → Cu2+ + Fe2+ + 2 S0 + 4 H2O
Vzniklá elementární síra tvoří na povrchu elektrody povlak, který brání galvanickému rozpouštění minerálů. Bakterie Acidithiobacillus ferrooxidans tuto síru oxidují na síranový aniont, odstraňují tak nerozpustný povlak síry a umožňují další pokračování galvanického rozpouštění.
Průběh technologie biologického loužení pomocí thionových baktérií je možné rozdělit do tří fází:
a) vlastní loužení
b) získávání kovů nebo více kovů z výluhu
c) regenerace loužícího roztoku
Proces bakteriálního loužení sulfidických rud znázorňuje obrázku 31.
Při vlastním loužení je extrakční roztok o pH 2,3 – 2,5, obsahující bakteriální kulturu v aktivní (exponenciální) fázi růstu, železo ve formě trojmocného iontu Fe2(SO4)3 a kyselinu sírovou, přivedem do kontaktu s louženou rudou. V loužené zóně dochází k oxidaci sulfidických minerálů bakteriemi a síranem železitým, k bakteriální oxidaci vznikající síry na kyselinu sírovou a k rozpouštění sulfidických nebo oxidických minerálů v kyselině sírové. V této fázi loužení je zapotřebí velkého množství loužícího roztoku a kyslíku v loužené zóně. Výluhy obsahují síran extrahovaného kovu, síran železnatý a železitý a bakteriální populace vyplavené z loužené zóny. Extrahovaný kov se získá z roztoku v technologické části provozu některými vhodnými metodami (cementace, elektrolýza, extrakce, iontová výměna, srážení a jiné).
Vzhledem k tomu, že výluh i odpadní roztoky po oddělení kovů obsahují kovy v toxických koncentracích, je nutno provádět loužení v uzavřeném cyklu nebo odpadní vody před vypouštěním do toku dočišťovat. Po odstranění předmětného kovu z výluhu se provádí regenerace loužícího roztoku pro jeho použití v dalším cyklu loužení. Přidává se kyselina sírová pro snížení pH na požadovanou aciditu a pomocí baktérií je dvojmocné železo oxidováno na trojmocné.
Technologie postupu bakteriálního loužení:
1. Příprava rudného materiálu (drcení, mletí), gravitačně, flotačně, aj.
2. Bakteriální loužení - přechod kovu z rudného materiálu do roztoku
3. Oddělení tuhé a kapalné fáze (zahušťování, filtrace, promývání)
4. Příprava roztoků k vydělení z nich čistých sloučenin nebo kovů
5. Vydělení čistých sloučenin nebo kovů (extrakce, cementace, elektolýza atd.)
6. Regenerace cirkulujících (vratných) roztoků se zachováním v nich bakterií a nebo pěstování bakterií v odděleném uzlu
|
|
|
Obrázek 32: Schéma bakteriálního loužení rud |
První informace o vlastnostech mikroorganismů a jejich schopnosti oxidovat rudy se získávají v laboratoři. Jde většinou o diskontinuální postupy.
Loužení v baňkách (bioreaktorech) za stacionárních podmínek
Rozemletá ruda přichází do styku s médiem a bakteriemi v laboratorních baňkách. Proces probíhá za volného přístupu vzduchu na klidném místě po dobu několika týdnů až měsíců. Provzdušňování roztoku je velmi špatné a tudíž i aktivita baktérií je nízká.
Loužení v baňkách (bioreaktorech) za neustálého míchání
Mícháním jemně rozemleté rudy s médiem a baktériemi v laboratorních baňkách se dosáhne lepšího nasycení suspenze vzduchem. Promíchávání se uskutečňuje probubláváním vzduchu, magnetickým míchadlem nebo třepáním.
Perkolátory
Perkolátory jsou nejčastěji používaná zařízení pro laboratorní studium bakteriálního loužení (obrázek 33). Perkolátor pojme 200 - 600 g rudy a 100 - 600 ml roztoku. Perkolátor sestává ze skleněného válce uzavřeného ve spodní části perforovaným porcelánovým dnem nebo skleněnou fritou. Na dno je do 1/2 až 2/3 nasypána rozemletá ruda. Pod tlakem je do postranní trubice vháněn vzduch obohacený oxidem uhličitým, který strhává ze spodní části baňky loužící roztok a vynáší ho do horní části perkolátoru. Rychlost perkolace závisí na pórovitosti materiálu, obsahu jílů, výšce náplně a tlaku vzduchu.
|
|
|
Obrázek 33: Perkolátor |
Loužící kolony
Jsou několik metrů dlouhé válce o obsahu až 200 tun rudy. Podobají se perkolátorům, jsou vyrobené ze skla, plastické hmoty nebo oceli. Cirkulace loužícího roztoku a jeho promíchávání se vzduchem a CO2 je jako u perkolátoru. Malé laboratorní kolony jsou používány pro modelování provozních podmínek loužení hald. Jsou vybavené automatickým vybavením pro měření O2, CO2, pH a teploty.
Loužení odvalů a hald
Při provozním loužení je loužící roztok vháněn na vrchol hald, kde je rozváděn kanálky do husté sítě rybníčků. Účinnější je zkrápění z perforovaných trubek nebo speciálním systémem sprch. Aplikuje se též injektování pomocí vrtů, což umožňuje občas nahradit roztok stlačeným vzduchem, obohaceným o CO2 pro provzdušnění hald. Roztok protéká rozdrcenou rudou, ze které se ve spodní části haldy sbírá a odvádí se k extrakci a odtud do oxidační nádrže, kde za přítomnosti Acidithiobacillus ferrooxidans se Fe2+ mění na Fe3+ . Regenerovaný roztok se vede zpět na vrchol haldy. Celý proces loužení je znázorněn na obrázku 34.
Odvaly jsou podle terénu umístěné na nepropustný podklad (vrstva jílu, asfaltu, cementu, gumy, plastické hmoty). Chudá ruda je navážená na hromadu tvaru komolého kuželu. Většina starých hald má výšku cca 200 m, šířku na vrcholu 80 m a paty okolo 200 m a obsahuje 50 000 - 300 000 tun rudy. V současnosti se haldy stavějí do tvaru prstovitých výběžků, jsou dlouhé několik stovek metrů a vysoké jen 10 - 15 m. Výhodou je to, že velká povrchová plocha umožňuje dobré provzdušnění a snížení rizika vzrůstu teploty uvnitř haldy nad požadovanou hranici.
|
|
|
Obrázek 34: Schéma procesu loužení sulfidických rud zkrápěním hald |
Loužení v nádržích
Povrch náplně je zkrápěn loužícím roztokem, který prosakuje rudou a ze dna je po obohacení odváděn. Většinou se nádrže spojují do baterií v protiproudém uspořádání, kde již částečně nabohacený roztok je použit k loužení čerstvé rudy, kdežto čerstvý roztok reaguje s již částečně vylouženou rudou. Patří sem loužící tanky typu PACHUCA (válce o průměru 3 m s výškou 12 m a s kuželovým dnem). Ve středu válce je umístěná vertikálně na obou koncích otevřená trubice, kterou se vhání vzduch pod tlakem a jeho bubliny udržují suspenzi ve vznosu.
Loužení in situ (loužení na místě)
Tento způsob je aplikován v opuštěných zavalených nebo zatopených uranových a měděných dolech. Z vrchu důlních děl je veden hlavní a nejhlubší vrt procházející horizontálními chodbami, které jsou situované pod rudným tělesem. Pro dávkování loužícího roztoku je provrtáno několik zavlažovacích sond. Roztok prosakuje rudním tělesem, extrahuje z rudy kovy a obohacený se čerpá na povrch ze dna nejhlubšího vrtu. Obdobně mohou být louženy „in-situ“ staré haldy.
„In situ“ loužení je vhodné pro sedimentární ložiska, které mají dostatečně velký součinitel filtrace pro průnik loužícího roztoku. Zvýšení extrakční schopnosti u nepropustných hornin se dociluje narušením jejich celistvosti střelnými pracemi s velkou silou výbuchu. Schéma podzemního loužení je znázorněno na obrázku 35.
|
|
|
Obrázek 35: Schéma podzemního loužení rud in-situ |
Moderní průmyslové využití biohydrometalurgie začalo bioloužením mědi z podřadného, těženého materiálu. Kennecott Copper Corporation úspěšně používala tento postup od 50.let minulého století. Tohoto příkladu následovaly i jiné důlní provozy v dalších zemích. V dnešní době zůstává odvalové bioloužení velmi nenákladným procesem získávání mědi z hornin, které nemohou být ekonomicky zpracovány jinou metodou. I přes komerční úspěch odvalového bioloužení byla u příslušného loužícího procesu vyvinuta velmi malá snaha o zlepšení mikrobiologické složky. Uplatnění biohydrometalurgie i v těžbě jiných kovů nastalo až v polovině 80.let 20.století, kdy byl uveden do provozu první závod na předúpravu zlatonosného koncentrátu. Současné postupy bioloužení mědi a předúpravy zlata jsou prováděny za použití podpůrných mikroorganismů.
Prognózy budoucího průmyslového využití biohydrometalurgie byly v roce 1986 formulovány na workshopu „Biotechnologie v důlním průmyslu, rafinaci kovů a zpracování fosilních paliv“ [15]. Tehdy jeden autor, J.F.Spisak, konstatoval, že využití biotechnologií v důlním průmyslu jsou v „období mládí“. Předpokládalo se, že biohydrometalurgie bude potřebovat 10 až 15 let na rozvoj pracovních postupů, praktické provedení a zkoušky v praxi. Spisak prohlásil, že největšími překážkami v aplikaci mikrobiálních postupů v těžebním průmyslu bude získat od vedení podniku souhlas a finanční prostředky. V roce 1986 nebyla technická proveditelnost podstatnou překážkou. Další účastník onoho workshopu, V.I.Lakshmanan, předpovídal průmyslové biohydrometalurgii podobnou budoucnost. Tvrdil, že rozvoj využití biohydrometalurgie v důlním průmyslu bude omezováno konzervativním přístupem k novým technologiím. Lakshmanan předpokládal, že přijetí bude pomalé a bude záležet na představivosti a míře risku ze strany nejvyššího vedení podniků. Dnes zůstávájí odhady pánů Spisaka a Lakshamana částečně opodstatněné. Avšak, zájem nejvyššího vedení mnoha podniků o biohyhrometalurgii stále roste, jelikož je na ni pohlíženo jako na důležitou komerční technologii budoucnosti, a to díky své jednoduchosti, nízkým nákladům a použitelnosti u chudých rud.
Získávání mědi
Rané průmyslové využití technologie bioloužení zpracovávalo podřadnou mědinosnou rudu v odvalech. Nedávná aplikace této technologie využívá upravené bioloužící haldy. Tyto postupy jsou uplatňovány v zemích jižní hemisféry. Stojí za zmínku, že průkopnické práce v Severní Americe nebyly dotaženy až k průmyslovému využití. Od roku 1980 bylo uvedeno do provozu deset bioloužících provozů na měď (tabulka 5).
Výborným příkladem současného průmyslového využití bioloužení je provoz Quebrada Blanca v severním Chile. Tento bioloužící závod se nachází na Alti Plano ve výšce 4 400 m.n.m., což zároveň vyvrátilo pochyby některých provozovatelů, že loužící bakterie nejsou schopny činnosti kvůli studeným teplotám, malému množství kyslíku a parciálnímu tlaku díky vysoké nadmořské výšce. V Quebrada Blanca se denně podrtí 17 300 tun sulfidické rudy 100% pod 9 mm, pokropí kyselinou sírovou a vrství se do tvaru 6 – 6,5 m vysokých hald. Činnost bakterií podporuje provzdušňování zajištěné řadou přívodů vzduchu, které jsou instalovány pod haldou a nízkotlakými ventilátory. K udržení adekvátních úrovní amoniaku (10 – 20 mg/l) a fosfátů (30 – 40 mg/l) pro činnost bakterií jsou do vyluhovacího roztoku přidávány živné látky. Monitoring bakteriálnho procesu zahrnuje respirační měření na místě. Bioloužící proces v Quebrada Blanca dokazuje úspěšnou „evoluci“ biohydrometalurgie v těžebním průmyslu. Konstrukce závodu v Quebrada Blanca a další podobné provozy začlenily bakteriální požadavky tohoto procesu. V současné době se v průmyslových provozech aplikují vědecké poznatky z oblasti zlepšování činností bakterií.
|
Tabulka 5: Průmyslové závody provozující loužení mědi na biohaldách |
|||||||||||||||||||||||||||||||||
a≈1,2 milionů tun rudního tělesa |
Předúprava zlata
Zlato se v přírodě vyskytuje v různých formách. Jako ryzí zlato například v podobě zlatinek nebo zlato vázané ve slitinách s jinými kovy. Podle výzkumů za použití transmisní elektronové mikroskopie (TEM) a Mössbauerové spektroskopie se zjistilo, že zlato je vázané také do krystalické mřížky některých sulfidických minerálů jako jsou například pyrit, arzenopyrit, chalkopyrit, antimonit nebo sfalerit. V této formě výskytu zlata v rudách (rezistentní zlaté rudy) jsou metody loužení v kyanidových solích nebo thiomočovině jen málo účinné, poněvadž i při velmi jemném mletí se zlato uzavřené v krystalických mřížkách minerálů nemůže dostat do kontaktu s rozpouštědlem.
V takových případech se používají metody umožňující rozklad zlatonosných sulfidů a to:
1. pražení (2 hodiny při teplotě 450 ºC a 2 hodiny při teplotě 650 ºC)
2. loužení za tlaku (autoklávy s mechanickou agitací 2-3 hodiny při teplotě 180 ºC)
3. biochemická oxidace
Pro předzpracování sulfidického zlatonosného koncentrátu biooxidací, před následující kyanizací nebo loužením v thiomočovině, bylo uvedeno do provozu šest závodů (tabulka 6). Tyto závody používají velké, provzdušněné, míchané reaktory na biooxidaci zlatonosných pyritů a arzenopyritů pomocí mezofilních bakterií rodů Acidithiobacillus, Leptospirillum, termofilních bakterií rodu Sulfolobus nebo jejich směsných kultur, popřípadě jiných kultur izolovaných a speciálne šlechtěných pro tyto účely, avšak blíže nespecifikovaných z důvodu obchodního tajemstvi.
Závod Youanmi v Austrálii, který je momentálně uzavřen kvůli nízkým cenám zlata a vysokým nákladům na těžbu, používá technologii BacTech, která je postavena na směsi 3 morfologicky odlišných bakteriálních druhů získaných izolací z místních půdních kultur a označovaných jako bakterie mírně termofilní. Tyto bakterie jsou autotrofní, oxidují dvojmocné železo a síru a na rozdíl od Acidithiobacillus ferrooxidans dokáží oxidovat i As3+ na As5+, což usnadňuje vázat As po rozložení arzenopyritu na tuhou stabilní sraženinu arzeničnan železitý. Biooxidace probíhá při teplotách mezi 30°C a 55°C. Tato směsná kultura bakterií byla adaptovaná až na pozoruhodnou hodnotu 25 g/l As v roztoku. Při oxidaci arzenopyritu na 55% se zvýšila výtěžnost Au z 35% na více než 90%.
Bac Tech kultura je podrobena rozsáhlým laboratorním zkouškám nejen na rezistentní zlaté rudy, ale i na rudy mědi a niklu.
Dalších pět závodů na zpracování sulfidických zlatonosných koncentrátů používá postup BIOX, což je směsná kultura Acidithiobacillus a Leptospirillum, která je aktivní mezi 40°C až 45°C. Předúprava biooxidací v reaktorech uzavřených v nádrži se průmyslově využívá jen u vysoce kvalitních flotačních koncentrátů. Biooxidace celé rudniny nemůže obecně ustát spojené náklady na energii na zajištění provzdušňování.
|
Tabulka 6: Průmyslové biooxidační závody na úpravu flotačních Au-koncentrátů |
|||||||||||||||||||||
a Závod Fairview byl uveden do provozu v roce 1986 a v roce 1991 expandoval na 35 tun/den |
Předúprava méně kvalitní rudniny biooxidací může být provozována v haldách, podobně jako u bioloužení mědi. Tento postup využívající biohald stále čeká na širší průmyslové využití, i když Newmont Gold Company prokázala použitelnost předúpravy biooxidací v haldách v praxi, a to u velkých předváděcích hald. Biooxidace se provádí u rudniny podrcené na 100 % pod 12,7 mm. Haldy jsou větrány a předúprava se provádí v časových úsecích dlouhých až 270 dní. Oxidovaná ruda je pak z oblasti předúpravy přemístěna, neutralizována a loužena. Výtěžnost zlata se pohybuje mezi 60 a 80% obsažených hodnot, v závislosti na mineralogii a velikosti použitých zrn. K předúpravě v haldách biooxidací se většinou přistupuje, pokud je ruda méně kvalitní.
Současné uzavření
závodu na zpracování Au-rudniny biooxidací v Youanmi a zpoždění ve spuštění
průmyslového procesu předúpravy biooxidací v haldách v Newmontu se odráží
na cenách zlata. I když jsou inovační biohydrometalurgické postupy, jako loužení
v biooxidačních haldách, relativně nízkonákladové, jejich realizace ve velkém
měřítku vyžaduje značný kapitál. Některé společnosti možná nebudou ochotny takto
investovat,
a to v dobách nízkých cen za kovy a počkají s implementací, až nastanou
příznivější ekonomické podmínky.
Získávání jiných kovů
V současné době se biohydrometalurgie aplikuje v průmyslovém měřítku k loužení mědi a předúpravě zlatonosných rud a koncentrátů. Ve velkém měřítku byla také předvedena funkčnost bioloužení u uranu. Bioloužení a bioúprava široké řady základních a platinových kovů mají značný potenciál. Mikrobiální loužení sulfidů základních kovů – Co, Ga, Mo, Ni, Zn a Pb – bylo realizováno v poloprovozních podmínkách. Sulfidické minerály obsahující platinové kovy (Pt, Rh, Ru, Pd, Os a Ir) mohou být předupravovány mikrobiálně. Další možné průmyslové využití bioloužení se jeví u základních kovů jako jsou nikl a kobalt.
Billiton vyvinul metodu BioNIC®, což je biohydrometalurgická technologie k získávání niklu z méně kvalitních sulfidických rud. Tato technologie je založena na postupu Goldfields BIOX® k předúpravě zlatonosných koncentrátů biooxidací. Poloprovozní testy bioloužení niklu z pentlanditu v komplexním sulfidickém koncentrátu prokázaly účinnost smíšené kultury Acidithiobacillus ferrooxidans, Acidithiobacillus thiooxidans a Leptospirillum ferrooxidans. Bioloužící část postupu je úspěšná, ale komerční využití bude také záviset na selektivní výtěžnosti niklu z loužicího roztoku. Tento aspekt je možný při uplatnění konvenčních metalurgických metod iontové výměny či extrakce rozpouštědly. Biotechnolog si musí být vědom skutečnosti, že prokázaný účinek mikroorganizmů je pouhou součástí průmyslového postupu a je třeba vše posoudit, včetně ekonomické stránky, před tím než je závod postaven.
Bioloužení kobaltu z pyritových koncentrátů je už velmi blízko průmyslovému využívání, možný je rok 2005. Návrhy projektu Kasese v dole Kilembe v Ugandě už byly dokončeny. K bioloužení kobaltu bude použito inokulum mezofilních bakterií oxidujících železo v systému míchaných reaktorů. Slibné pokroky v bioloužení kobaltu a niklu jsou předzvěstím průmyslového využití biohydrometalurgického zpracování základních kovů, jiných než měď.
Kušnierová a kol.(2002) uvádí aplikační možnosti mikroorganismů v minerálních biotechnologiích. V tabulkách 7 a 8 je uveden přehled mikroorganismů při loužení vybraných kovů.
|
Tabulka 7: Aplikační možnosti mikroorganismů v minerálních technologiích |
||||||||||||||||||||||
|
||||||||||||||||||||||
|
Tabulka 8: Schopnost mikroorganizmů loužit některé kovy |
||||||||||||||||||||||||||||||||||||||||||||||||
|
Roztok užitkové složky – výluh, který získáme z procesu loužení, je zpravidla velmi znečištěný jemnými zrníčky jaloviny z louženého materiálu. Tyto tuhé částečky je třeba před srážením z výluhu odstranit. Hlavní technologické postupy, které se při čištění výluhu uplatňují, jsou běžně používané technologické postupy zahušťování a filtrace. Pokud mají částice znečišťující výluh koloidní rozměry, intenzifikuje se jejich sedimentace koagulací anorganickými elektrolyty nebo flokulací organickými makromolekulárními sedimentačními reagencemi.
Na zahušťování se používají diskontinuálně pracující sedimentační nádrže převážně čtyřúhelníkového půdorysu nebo kontinuálně pracující kruhové zahušťovače s mechanickým nepřetržitým shrnováním sedimentu k vynášecímu otvoru ve středu kuželovitého dna. Na rozdělení tuhé fáze od kapalné se v chemické úpravě používají různé konstrukce podtlakových i přetlakových filtrů.
Protože procesy zahušťování a filtrace znamenají v hydromentalurgii značné náklady zatěžující zpracování, projevuje se v poslední době snaha získat kovy přímo ze rmutů. Jako příklad lze uvést využívání iontoměničů nebo kapalných extrakčních činidel, jako je tomu u procesu Solvent in Pulp. Při zpracování výluhů s obsahem mědi je doporučen způsob jejího vycementování přímo ze rmutu. Po aktivaci se pak prahová měď odděluje flotací. Uvedený proces Solvent in Pulp šetří nejen značně vysoké náklady za usazování, ale umožňuje i mnohonásobné zmenšení objemu kyselých roztoků potřebných k loužení. To se pak projeví ve snížené spotřebě extrakčních organických činidel.
Iontoměniče jsou tuhé makromolekulární látky nerozpustné ve vodě a běžných rozpouštědlech skládající se z pevné kostry – matrice, kterou tvoří elektricky nabité iontové skupiny a v mezerách se volně pohybují ionty opačného náboje. Ionty, které chceme zachytit na iontoměniči jsou pohyblivé a elektricky vyvažují náboj iontových skupin matrice. Jsou nahrazovány ionty se stejným nábojem, které obsahují funkční skupiny matrice.
Iontovou výměnu můžeme zjednodušeně charakterizovat jako výměnu mezi ionty tuhé fáze (iontoměniče) za ekvivalentní množství iontů obsažených v roztoku, se kterým přichází tuhá fáze do styku. Při tomto procesu se tuhá fáze podstatně nemění.
Jsou známé mnohé přirozené i syntetické látky, které mají schopnost vyměňovat ionty, avšak v technické praxi jsou obzvláště významné měniče iontů na bázi syntetických pryskyřic (obrázek 36) a méně pak minerální měniče iontů.
|
|
|
Obrázek 36: Matrice iontoměniče vyrobená na bázi syntetické pryskyřice |
Podle charakteru a vlastností účinných funkčních skupin se iontoměniče (ionexy) dělí na:
měniče kationtů (katexy), s kyselými funkčními skupinami,
měniče aniontů (anexy), se zásaditými funkčními skupinami,
amfoterní, s oběma druhy funkčních skupin.
Pokud ionexy obsahují jediný druh funkčních skupin, jsou monofunkční, pokud obsahují více funkčních skupin jsou polyfunkční.
Živicové (pryskyřicové) měniče iontů jsou uměle připravované ionexy s výbornou chemickou i mechanickou stálostí. Jejich rozvoj souvisí s vývojem makromolekulární chemie. Základem (matricemi) těchto iontoměničů jsou vysokomolekulární prostorové sítě velkých uhlovodíkových řetězců. Na této matrici jsou pevné, kovalentní vazbou vázané, funkční skupiny schopné elektrolytické disociace a tedy i výměny iontů.
Funkční skupiny katexů:
R - SO3-H+ sulfonová
R - COO-H+ karboxylová
R – PO32-H22+ fosfonová alkyl(aryl) fosforečná
R – AsO32-H+ arzenová alkyl (aryl) arzeničná
Funkční skupiny anexů:
R – NH3+CI- primární aminová
R2 - NH2+CI- sekundární aminová
R3 - NH+Cl- terciární aminová
R4 - N+OH- kvartérní amoniová
Katexy
Základem katexu je plastová kulička, vyrobená například z polystyrenu a divinylbenzenu. Na takto získanou plastovou kuličku se vhodným způsobem navážou tzv. funkční skupiny jako například R-SO3-H+ (zbytek kyseliny sírové), které disociují v celém rozsahu pH (jsou silně kyselé). Funkční skupiny COO-H+ disociují jen v zásaditém prostředí (jsou slabě kyselé). Tyto skupiny jsou schopny vyměňovat vodík za různé ionty kovů, např. sodík, vápník, hořčík, draslík, železo apod.
V anexech v celém rozsahu pH disociují kvartérní aminy (silně zásadité), zatímco skupiny primárních, sekundárních a terciárních aminů disociují jen v kyselém prostředí (slabě zásadité). Matrice může být například kopolymer na bázi styrenu a divinylbenzenu.
Aminy jsou sloučeniny, které lze odvodit od amoniaku náhradou vodíkových atomů uhlíkovými zbytky (obrázek 37). Nahrazením jednoho atomu vodíku vznikají aminy primární R - NH2, nahrazením dvou atomů vodíku aminy sekundární (R)2NH a nahrazením tří atomů vodíku aminy terciarní (R)3N. Při vazbě čtyř radikálů vznikají kvartérní amoniové soli [(R)4N]+X-.
|
|
|
Obrázek 37: Strukturní vzorce aminů |
Moderní ionexy se vyrábějí ve formě kuliček s přibližnými rozměry 0,2 – 1,2 mm. K nejdůležitějším fyzikálně-chemickým vlastnostem ionexů patří výměnná kapacita. Je to množství iontů (hmotnostní nebo objemové), které je jednotka ionexu schopná vyměnit. Toto množství je určováno počtem tuhých iontů obsažených ve hmotě ionexu, protože jejich elektrický náboj musí být na každém místě měniče kompenzovaný nábojem pohyblivých iontů roztoku. Iontová výměna je proces přesně stechiometrický.
Ionex přednostně sorbuje:
ion s vyšším mocenstvím pro jeho vyšší elektrostatický náboj,
iont menšího průměru na základě mechanického síťového efektu,
ion, který ve větší míře tvoří s efektivními skupinami ionexu málo disociované soli nebo komplexy.
Rychlost výměny iontů závisí od rychlosti pronikajících iontů matricí tvořící ionex. Na rychlost difůze a tím i na rychlost výměny iontů má vliv struktura ionexu, hustota prostorové sítě základního polymeru a charakter i množství funkčních skupin.
Práce s ionexem probíhá v dynamických podmínkách. Přes určité množství ionexu A v koloně nebo na filtru protéká roztok výluhu B. Když sledujeme koncentraci iontů B v roztoku vytékajícím z kolony (filtrát) naplněné ionexem A a graficky znázorníme závislost koncentrace iontu B na objemu proteklého vzorku, dostaneme sorpční křivku (obrázek 38 a). Když obrátíme proces a budeme na ionex přidávat roztok elektrolytu A (u katexu např. HCl), bude nám ze živice vytěsňovat ion B. Tento proces se nazývá eluce, regenerace nebo desorpce. Příslušný průběh závislosti koncentrace iontu B v eluátu na objemu eluátu bude podle obrázku 38 b.
|
|
|
Obrázek 38 a,b: Sorpční a desorpční křivka ionexu |
Nejčastěji probíhá výměna iontů v iontoměničových kolonách, kde roztok protéká přes stabilní vrstvu ionexů. V praxi bývá většinou několik sorpčních kolon zapojených za sebou (obrázek 39). Na obrázku jsou znázorněny 4 kolony, ze kterých 3 pracují jako sorpční a ve čtvrté probíhá současně eluce (desorpce). Kolony se zapojují v pořadí cyklicky tak, že když se první přívodní kolona nasytí, přepojí se na desorpci a regenerovaná kolona po eluci pak začíná svoji činnost znovu jako sorpční kolona.
Na obrázku 40 je pak zobrazen poloprovozní katexový iontoměnič.
|
|
|
Obrázek 39: Řada iontoměničových kolon zapojených za sebou |
|
|
|
Obrázek 40: Katexový iontoměnič |
Druhý způsob iontové výměny je promíchávání roztoku (nebo přímo rmutu) iontoměničem. Tento způsob se využívá v chemické úpravě rud zlata, uranu, molybdenu apod. Vyžaduje mechanicky pevnější iontoměnič zrnitosti 0,5 – 1,5 mm a mletí rudy na jemnost pod 0,1 mm. Zahuštění rmutu je 1:1. Rmut se s iontoměničem promíchává pneumaticky. Moderní výměnný iontoměničový systém, který byl vyvinutý na zdokonalení výměny iontů (pro extrakci uranu), je schematicky znázorněn na obrázku 41.
|
|
|
Obrázek 41: Schéma protiproudé iontoměničové kolony 1 - díl kolony, 2 – přívod iontoměniče, 3 – vrstvy iontoměniče, 4 – dírkované přepážky, 5 – výpusť iontoměniče, 6 – odtok filtrátu, 7- přívod roztoku |
Roztok stoupá v koloně zdola nahoru rychlostí, která fluidizuje lůžko iontoměniče a zabraňuje přepadavání zrn iontoměniče otvory v dírkovaných přepážkách. Periodicky se přívod roztoku přeruší a část iontoměniče propadne z vyšších oddělení do nižších a část z nejnižšího oddělení (obohacený iontoměnič) se vypouští, aby se eluoval.
Základní předpoklady
Voda vstupující na ionexy musí být čirá, zbavená mechanických nečistot, suspendovaných látek a koloidních částic. Ionexové filtry nemají sloužit jako mechanický filtr. Při předřazeném čiření nebo dekarbonizaci vápnem nemá na ionexech docházet k dobíhání srážecích reakcí.
Vstupní voda na ionexy nesmí obsahovat olej (například při změkčování kondenzátu) a volný chlór v množství vyšším než 0,2 mg/l. Má obsahovat co nejmenší množství neiontového železa a hliníku.
Většina závad v provozu ionexových filtrů je způsobena nedokonalou nebo špatně provozovanou předúpravou a nedostatečným, popřípadě málo intenzivním praním.
Pracovní cyklus iontoměniče můžeme rozdělit na:
pracovní fáze (dochází k výměně iontů)
praní (odstranění nečistot zachycených ionexem a nakypření ionexu po pracovním období)
regenerace (převedení ionexu pomocí regeneračního činidla zpět do činné formy – HCl, NaCl, NaOH)
vymývání (odstraňování z ionexu přebytečný regenerační roztok)
Na základě výsledků experimentů bylo sestaveno pořadí příbuznosti různých iontoměničů k iontům v roztoku.
Pro katexy:
Na+ ‹ Ca2+ ‹ Al3+ ‹ Th4+
Pro sulfokatexy:
Li+ ‹ H+ ‹ Na+ ‹ NH+ ‹ K+ ‹ Rb+ ‹ Cs+ ‹ Ag+ ‹ Hg2+ ‹ Ca2+ ‹ Sr2+ ‹ Ba2+ ‹ Al3+ ‹ Fe3+
Pro katexy s karboxylovou skupinou:
Cs+ ‹ Rb+ ‹ K+ ‹ Na+ ‹ Li+ ‹ Mg2+ ‹ Ca2+ ‹ H+
Pro amidoacetálové katexy:
K+‹‹ Na+ ‹‹ Li+ ‹‹ Ba2+ ‹ Mg2+ ‹ Ca2+ ‹ Mn2+ ‹ Fe2+ ‹ Co2+ ‹ Zn2+ ‹ Cd2+ ‹ Ni2+ ‹ ‹ Pb2+ ‹ Cu2+ ‹ NO22+
Pro silně zásadité anexy s kvarterními amoniovými skupinami:
F- ‹ OH- ‹ Cl- ‹ NO- ‹ CN- ‹ Br- ‹ NO3- ‹ HSO4- ‹ SCN- ‹ ClO- ‹ MoO42- ‹ CrO42- ‹ SO42- ‹ PO43- ‹ AsO43-
V hydrometalurgii se pod pojmem kapalinová extrakce (extrakce organickými rozpouštědly) rozumí převádění solí kovu z vodného roztoku do kapalné organické fáze nemísitelné s vodou. Účelem převedení kovu z vodné fáze do organické může být buď jeho získání v čisté formě nebo jeho odstranění z roztoku, jestliže je tento kov nečistotou.
Látka, která nás zajímá (kov nebo jeho sloučenina), reaguje s extrakčním činidlem za tvorby chemické látky, která je rozpustnější ve fázi organické než ve fázi vodné. Vzniklá sloučenina tedy přechází do organické fáze.
Následnou reextrakcí se pak získá extrahovaný kov nebo jeho sloučenina zpět do vodného roztoku. Koncentrace látky v „novém“ vodném roztoku bývá často 10-100 krát vyšší než v původní vodné fázi a organický roztok se vrací na další extrakci buď přímo nebo po regeneraci. Princip kapalinové extrakce je na obrázku 42.
|
|
|
Obrázek 42: Principiální schéma kapalinové extrakce |
Základní technologické termíny používané v technologii extrakce organickými rozpouštědly:
Extrakční činidla – rozpustné v organických kapalinách, jsou to organické kyseliny, alkoholy, estery, ketony, aminy a jiné (obrázek 43).
Organická ředidla – petrolej, xylol, lehký olej. Organické ředidlo zmenšuje zahuštění, viskozitu extrakčního činidla a jeho ztráty. V případě použití aminu jako
extrakčního činidla se jako ředidlo používá petrolej s triethylenglykolem nebo oktylalkoholem. Jiná ředidla jsou diizopropylester, polyalkylbenzol apod.
Vysolovadlo - neorganická látka (obvykle elektrolyt), vylepšující ukazatele extrakce - obvykle přispívá k tvorbě extrahovatelných komplexů. Vysolovací reagence podporuje dehydrataci iontů extrahovaného iontu a jeho solvataci molekulami extrakčního činidla.
Extrakt - organická fáze po extrakci.
Rafinát - vodní fáze po extrakci (odpad).
Reextrakt – vodní fáze získaná reextrakcí kovu z extraktu do vodné fáze (odpad).
V průmyslovém rozsahu se začala poprvé používat kapalinová extrakce při získávání dusičnanu uranylu na výrobu kovového uranu pro palivové články.
Po tomto úspěchu se extrakce vodnými rozpouštědly podstatně rozšířila i na jiné kovy.
|
|
|
Obrázek 43: Složení organické fáze |
Při výběru extrakčního činidla se zohledňuje více faktorů. Nejvýznamnější jsou tyto:
a) dostatečně velká selektivita vůči extrahovanému kovu,
b) rychlé dosáhnutí extrakční rovnováhy,
c) stálost extrakčního činidla vůči používaným chemikáliím,
d) extrakční činidlo nesmí tvořit těžko oddělitelné emulze a oddělení organické a vodní fáze má být rychlé a úplné,
e) dostupná cena extrakčního činidla,
f) hořlavost, toxicita a extrakční kapacita extrakčního činidla.
Hlavní oblast používání extrakce organickými rozpouštědly se používá v průmyslu uranu, india, thália, germánia, teluru, wolframu, mědi, dále při odddělení Co a Ni, Ta a Ni, Zr a Hf, Mo a Re.
Podle způsobu extrakce, popřípadě podle reakcí probíhajících při extrakci, rozeznáváme čtyři skupiny:
Jednoduché fyzikální rozdělení – do této skupiny patří fyzikální (mechanická) extrakce symetrických kovalentních molekul, jejichž rozpustnost je řádově vyšší v organických rozpouštědlech než ve vodě.
Extrakce na principu výměny kationtů - do této skupiny patří extrakce kationtů kovů organickými kyselinami, jejich solemi a chelátotvornými reagencemi.
Příkladem extrakce tohoto typu může být extrakce kationtů kobaltu Co2+ sodným mýdlem mastné kyseliny
2 RCOO Na+ + Co2+aq → (RCOO)2Co + 2 Na+aq
Extrakční činidla patřící do této skupiny jsou soli mastných kyselin se 7 až 9 uhlíky v radikálu, alkylfosforečné kyseliny a jejich soli. Organické kyseliny se obvykle zřeďují v inertním rozpouštědle (např. v petroleji).
Karbonátové a naftenové kyseliny vytvářejí s extrahovanými kovy soli (tzv. mýdla, rozpustná v použitém rozpouštědle). Používají se směsi karboxylových kyselin C7 – C9, popř. C10 – C16. Jejich selektivita je závislá na pH prostředí. Změnou pH se může dosáhnout poměrně ostrého oddělení jednotlivých kovů (obrázek 44). Můžeme je použít při oddělení některých kovů, např. Ni, Co, Cu z vodných roztoků a jejich solí (síranů nebo chloridů).
Karbonové kyseliny se v praxi používají na zpracování zředěných roztoků síranu měďnatého před elektrolýzou. Využívá se skutečnost, že extrakcí můžeme i z velmi zředěných roztoků připravit roztok mědi vhodný pro elektrolýzu podle obrázku č. 45.
Ze skupiny komplexotvorných činidel uvedeme např. Di-2-etylhexylfosforečnou kyselinu (D-2-EHFK), která v slabě kyselém prostředí extrahuje kationty mechanizmem katexové výměny iontů.
|
|
|
Obrázek 44: Závislost stupně extrakce dvojmocných kovů karboxylovými kyselinami na pH |
|
|
|
Obrázek 45: Schéma extrakce zředěného roztoku CuSO4 před následnou elektrolýzou. |
Protože má ve struktuře polární skupinu P=0, ve které kyslík je donorem elektronů, může dojít k doplňkovému připojení molekul kyseliny ke kationtu, přičemž se tvoří komplex. Příkladem vytvoření takovéto komplexní soli může být extrakce molybdenitových kationtů MoO22+
MoO22++ 2 (HR2PO4)2 → MoO2(R2PO4)2 2 HR2PO4 +2 H+
Strukturní vzorec vzniklé komplexní soli je:
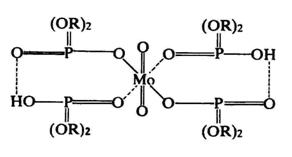
V moderních technologiích chemické úpravy rud se velmi rozšiřuje používání extrakčních činidel s oximovými a hydroxyoximovými skupinami:
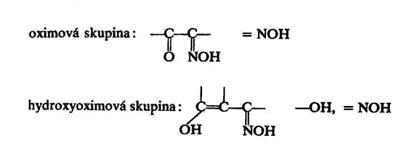
3. Extrakce na principu výměny aniontů – představiteli této skupiny jsou aminy. V hydrometalurgii používané aminy mají radikály s počtem uhlíků 7 – 9.
Příkladem je TOA (trioktylamin – (C8H17)3N (terciérní amin).
R3N + HNO3 → R3NH . NO3
Dusičnan TOA je schopný vyměnit svůj anion za anion obsahující kov, např. rhenium:
R3NH . NO3 + (ReO4)- → R3NH. ReO4 + (NO3)-
Reextrakce se provádí čpavkem.
4. Extrakce neutrálními extrakčními činidly – k neutrálním extrakčním činidlům patří ketony. Z technického hlediska je nejvýznamnější metylizobutylketon (MiBK) CH3COCH2CH(CH3)2 nebo strukturně:
Používá se na přepracování vyhořelého paliva a na izolaci plutonia. Příslušný technologický postup se nazývá REDOX. Z organické fáze se zředěnou kyselinou dusičnou reextrahuje uran a plutonium. Dalšími postupy se oba dva kovy rozdělí.
Do skupiny neutrálních činidel se dále zařazují alkylestery kyseliny fosforečné, především tri-n-butylester kyseliny fosforečné (tributylfosfát - TBF):
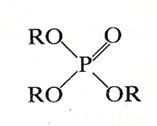
(kde skupinou R je CH3. CH2 . CH2. CH2 - ).
TBF se používá při zpracování dusičnanových roztoků aktinidů. V praxi se využívá při zpracování vyhořelého paliva jaderných reaktorů a na izolaci plutonia v procesu PUREX.
Proces extrakce nevodnými rozpouštědly se skládá ze dvou částí, a to z dispergované organické fáze ve vodném prostředí a z následujícího rozdělení vzniklé disperze v důsledku rozdílných hustot obou fází. Účinnost extrakčního procesu bude záviset na velikosti mezifázového povrchu tj. na stupni dispergace jedné fáze do druhé.
Přístroje, ve kterých probíhá dispergace organické fáze do vodného roztoku extrakcí a opětovné rozdělení obou fází, nazýváme extrakční kolony. Příkladem je etážová extrakční kolona (obrázek 46).
Je to vysoká nádrž válcovitého tvaru s přihrádkami. Lehčí kapalina – organická fáze se přivádí do spodní části a vodný roztok, který je specificky těžší, se přivádí do kolony v horní části. Těžší kapalina stéká po přihrádkách a na okrajích odkapává, čímž se dosáhne lepšího rozptýlení obou kapalin. Dispergace se zintenzivní zvýšením turbulence obou fází dodáním mechanické energie, buď ve formě pulzačního pohybu obsahu kolony, nebo mechanickým mícháním.
|
|
|
Obrázek 46: Etážová extrakční kolona |
Na extrakci kapalin s velkým mezifázovým napětím je vhodné použít stupňovité extraktory. Schéma oddělení mixéru a settleru je na obrázku 47. Obě dvě kapaliny se přivádí do mixéru zespod a promíchávají se lopatkovými míchadélky. Emulze se přečerpává do settleru, kde se v klidu oddělí těžká fáze od lehčí.
|
|
|
Obrázek 47: Stupňovitý extraktor a –mixér, b -settler |
V průmyslu se také často používá spojení několika extraktorů (3-4) pracujících za sebou.
Přednosti extrakce nevodnými rozpouštědly:
a) velká selektivita procesu,
b) možnost dosáhnutí vysokého stupně nabohacení už při jedné operaci,
c) v poměrně krátkém čase se může zpracovat velký objem vodného roztoku,
d) zařízení není příliš nákladné,
e) reextrakční roztoky mají daleko vyšší koncentraci extrahovaného kovu než jiné použité metody,
f) můžeme ji použít i při zpracování rmutu.
Nevýhody kapalinové extrakce:
a) poměrně vysoká cena extrakčních činidel,
b) určitá ztráta inertního rozpouštědla, způsobená rozpustností ve vodné fázi a také i nedokonalým oddělením obou fází.
Srážení kovů nebo jejich sloučenin z roztoku představuje jednu z hlavních operací v hydrometalurgickém zpracování surovin určující celkovou úspěšnost procesu.
V současné praxi se používají rozličné způsoby srážení kovů a jejich sloučenin. Výběr nejúspornějších metod, na které je třeba se v jednotlivých případech zaměřit, je mnoho a jejich výběr ovlivňuje např. charakter výchozí suroviny, přítomnost zdroje zásobování elektrickým proudem a cena elektrické energie, složení roztoků získaných při loužení, cena činidel, jakost (čistota) konečného produktu, bezpečnost a zdraví obsluhujících zaměstnanců.
Známé a používané jsou tyto způsoby srážení kovů nebo jejich solí z roztoku:
srážení stejnosměrným proudem (elektrolýza)
srážení kovů cementací
získávání chemických sloučenin kovů krystalizací
srážení hydrolýzou
srážení chemickými činidly
srážením ohřevem v destilátorech
srážení adsorpcí
Z uvedených způsobů srážení se odborníci v praxi nejčastěji přiklánějí k elektrickému srážení a cementaci.
Srážení z roztoku Cu, Zn, Cd, Mn se uskutečňuje převážně elektrolyticky. Cementační procesy můžeme použít při srážení zlata z kyanizačních roztoků, zinku, mědi atd. Kovy, získané cementací, mívají velmi nízký obsah vlastního kovu, a proto se musí před dalším použitím rafinovat. U mědi jde o rafinaci žárovou cestou s následující rafinační elektrolýzou. Ostatní způsoby srážení mají také svůj význam při chemickém zpracování rud, i když se méně používají.
Elektrolýza patří mezi elektrochemické procesy a používá se při získávání kovů z roztoků po loužení rud – elektrolytické srážení a na získávání čistých kovů ze surových kovů – elektrolytická rafinace.
Rafinace mědi se provádí ve dvou fázích. Nejdříve se měď přetavuje v plamenových pecích, kde se do roztavené měděné lázně dmýchá vzduch.Vzniká 99,7%-ní Cu která se odlévá do tzv. anodových bloků. Probíhá zde elektrolytická rafinace, kde anodou je surová Cu, katodou čistá Cu a elektrolytem je roztok CuSO4 a H2SO4. Surová měď se anodicky oxiduje a přechází do roztoku ve formě iontů odkud se vylučuje na katodě jako 99,99%-ní Cu.
Mezi oběma procesy jsou důležité tyto rozdíly:
a) elektrolytická rafinace se provádí s rozpustnými anodami, které se rozpouštějí působením proudu. Katody jsou při elektrolytickém srážení nerozpustné a jsou určeny pouze pro přenášení proudu v elektrolytu a na vybíjení kationtů.
b) Při elektrolytické rafinaci má elektrolyt téměř stejné složení. Při elektrolytickém srážení je elektrolyt ochuzen o ionty sráženého kovu, což si vynucuje kontinuální přidávání čerstvého roztoku pro udržení stálého složení elektrolytu.
c) Při elektrické rafinaci se přenášejí kationy kovu proudem od anody ke katodě. Velká část energie se vynakládá na překonání odporu elektrolytu. Energie vynaložená na rozpouštění kovů na anodě se kompenzuje energií, kterou uvolňují kationty, které se na anodě srážejí. Při elektrolytickém srážení se energie spotřebuje na rozložení solí těžkého kovu a je v porovnání s rafinací vysoká.
d) V průběhu elektrolytického srážení se na elektrodách tvoří plyny, které vyvolávají chemickou polarizaci s obrácenou elektromotorickou silou. Na její překonání je třeba větší množství energie.
e) Při elektrolytickém srážení se obvykle pracuje s méně čistými roztoky, než při elektrolytické rafinaci. Což vyvolává korozi a zpětné rozpouštění katodové usazeniny a dále se snižuje výtěžek na jednotku proudu a energie.
Elektrolytické srážení má velký význam v metalurgii mědi, zinku, kadmia a manganu, protože zaručuje získávání kovu nejvyšší čistoty.
Podstatou elektrolýzy jsou dva Faradayovy zákony:
Podle prvního Faradayova zákona je hmotnost látky m vyloučené při elektrolýze z roztoku na elektrodě přímo úměrná součinu intenzity elektrického proudu I a délce trvání t.
a dále pak
kde A je elektrochemický ekvivalent [Kg.C-1] (k = 1,0363 .10-2, M/ν – gramekvivalent, M je atomová nebo molekulová hmotnost, ν je valence). Gramekvivalent atomu nebo skupiny atomů je množství vyjádřené v gramech, které reaguje s 1,008 g vodíku.
H + ClI atomová hmotnost chlóru = 35,46 = gramekvivalent chlóru,
H2 +OII atomová hmotnost kyslíku = 16,00 = gramekvivalent kyslíku = 8,
H2 + SO4II molekulová hmotnost SO4 = 96,06, gramekvivalent SO4 = 48,03.
Druhý Faradayův zákon zní: při průchodu stejného množství elektrického proudu různými elektrolyty je množství látek vyloučené na elektrodách úměrné ekvivalentu hmotnosti těchto látek. Na vyloučení gramekvivalentu látky je třeba vydat vždy stejné množství elektrické energie a to 96 500 Coulombů.
Průběh rozkladu molekuly síranu měďnatého je znázorněn na obrázku 48.
|
|
|
Obrázek 48: Rozklad CuSO4 pomocí elektrolýzy |
Proudový výtěžek kovu (využití proudu):
Na vyloučení 1 gramekvivalentu látky je potřeba, aby elektrolytem proteklo větší množství elektřiny než 1 farad (1F = 96 500 C). To není porušením Faradayova zákona, ale vysvětluje se to vylučováním vodíku na katodě vedle kovu, oxidací a rozpouštěním katodové sraženiny, vedlejšími reakcemi, přítomností nečistot v elektrolytech a též rozptylem proudu a krátkými spojeními. Proudový výtěžek je poměr skutečného množství vysráženého kovu na katodě k množství, které by se mělo teoreticky získat při přechodu stejného množství elektrického proudu za stejných podmínek (bez vedlejších účinků). Proudový výtěžek pro různé kovy závisí na podmínkách elektrolýzy, pohybuje se ve velmi širokém rozsahu od 45 do 95%. Minimální elektromotorická síla, kterou je třeba dodat na vyloučení takového množství aniontů a kationtů na nerozpustných elektrodách, aby roztokem procházel elektrický proud, se nazývá rozkladné napětí elektrolytu.
Poměr množství energie Eo, které je teoreticky potřebné na elektrolytický proces, k množství E, které se skutečně spotřebuje na jednotku hmotnosti získaného kovu, se nazývá součinitel využití energie (B)
Struktura a fyzikální vlastnosti kovu, usazujícího se na katodě, má značný význam. Při elektrolytickém srážení kovů z roztoku i při elektrolytické rafinaci z kovů se snažíme získat husté sraženiny.
Každé usazování kovu na katodě začíná tvorbou krystalizačních jader, které se postupně rozrůstají. Pokud rychlost usazování iontů kovu převyšuje rychlost jejich přivádění, tvoří se hrubé krystalky kovů. V opačném případě se nárůst krystalů omezí nepřetržitou tvorbou nových krystalizačních středisek na povrchu kovu. V takovém případě dostaneme jemně krystalickou strukturu.
Se zvyšováním proudové hustoty se struktura vysráženého kovu stává jemnější, usazenina hustší, přičemž se na povrchu a okrajích katody objevují vyduté výrustky, které porušují usazeninu kovu.
Při elektrolytickém srážení kovu je důležitá koncentrace elektrolytu: čím je roztok koncentrovanější, tím bude krystalická sraženina hustší a jemnější. Snižováním koncentrace elektrolytu je sraženina méně hustá, až se nakonec z ní stane sypká, práškovitá hmota, která se lehce odlupuje z katody.
Přítomnost škodlivých příměsí v roztoku vyvolává při elektrolytickém srážení:
1. snížení výtěžku na jednotku proudu,
2. znečištění a zhoršení katodové usazeniny,
3. v některých případech zvětšení odporu elektrolytu, vyžadující zvýšení napětí v lázni.
Zařízení na elektrolytické srážení kovů z roztoku mají různou konstrukci, např. jsou dřevěné, poolověné, železobetonové s asfaltovou výstelkou, lázeň s otočnými katodami (pro Cd), s diafragmou (pro Mn). Elektrolyzér pro elektrolýzu mědi i niklu má tvar žlabu délky 20 m, šířky 1 m a hloubky 1,3 m. Železobetonový žlab může být vyložený olovem nebo asfaltem. Na bočních stěnách žlabu, na kterých jsou na dřevěných izolátorech vodiče stejnosměrného proudu, visí za sebou elektrody. Elektrody se zapouštějí a vytahují jeřáby. Anody jsou z olova, které obsahuje 7 až 8% antimonu. Katody, na kterých se sráží měď, jsou z měděných fólií (obrázek 49). Na elektrolytické srážení manganu se používá elektrolyzér s diafragmou, aby se izolovala katodová sraženina od kyselého akolytu, který by rozpouštěl kovový mangan. Diafragmy jsou plátna, které ohraničují prostor kolem anod.
|
|
|
Obrázek 49: Tvar elektrod (elektrolýza mědi) |
Když ponoříme jakýkoliv kov, např. zinek, do roztoku jeho solí, přejde do roztoku ve formě iontů
Mezi elektrodou a roztokem vznikne rozdíl potenciálů. Snaha kovu vysílat do roztoku své ionty se nazývá elektrolytický rozpouštěcí tlak. Při rozpouštění kovů přecházejí do roztoku jen kladné ionty a samotná elektroda se nabíjí záporně. V blízkosti elektrody se roztok nabíjí kladně a kladný náboj je elektrostaticky přitahován k záporně nabité elektrodě (obrázek 50). Tlak kladných iontů přitahovaných k elektrodě je osmotický tlak. Proto se ionty kovu nemohou velmi vzdálit od elektrody a v nejbližším okolí elektrody vytvoří roztok dvojitou vrstvu: záporně nabitou elektrodu a kladně nabitý roztok. Dvojitá vrstva překáží dalšímu přechodu iontů kovu do roztoku a nastává rovnováha rozdílu elektrolytického rozpouštěcího tlaku a osmotického tlaku.
Pokud je rozpouštěcí tlak elektrody větší než osmotický tlak iontů, bude mít elektroda záporný potenciál, při opačném poměru bude mít kladný potenciál.
Zn0 → Zn 2+ + 2e-
|
|
|
Obrázek 50: Schéma vytvoření elektrické dvojvrstvy v okolí zinkové destičky |
Pokud se rozpouštěcí tlak elektrody rovná osmotickému tlaku jeho iontů, potom se bude potenciál kovu, vzhledem k jeho roztoku, rovnat nule. Tento potenciál se nazývá normálový nebo standardní potenciál φ0.
Zinková elektroda ponořená do roztoku svojí soli má vzhledem k potenciálu vodíku záporný potenciál, měděná elektroda má kladný potenciál (viz. Beketovova řada elektrochemických napětí prvků). Tímto způsobem se rozlišují elektronegativní a elektropozitivní kovy. Tento jev si vysvětlujeme takto: Zinek je kov, který má relativně labilně vázané elektrony, které se ve styku s roztokem elektrolytu lehce ztrácí a přecházejí v iontové formě do roztoku.
Zn0 → Zn 2+ + 2e-
Kvalifikujeme ho jako méně ušlechtilý kov. Kovy s větší přitažlivostí k vazbě elektronů, např. měď, přecházejí do roztoku hůře. U mědi převládá tendence vysrážení iontů na neutrální měď. Takovéto kovy označujeme jako ušlechtilé.
Beketovova řada napětí
Pro většinu kovů se hodnoty normálových elektrodových potenciálů bezprostředně určili pomocí pokusů, avšak pro některé, např. pro alkalické kovy a kovy alkalických zemin, se vypočítaly teoreticky, poněvadž bylo obtížné toto stanovení provést pokusem.
Když uspořádáme normálové potenciály (φo) do řady podle jejich stoupajících algebraických hodnot, získáme řadu napětí. Za nulový potenciál se určil potenciál vylučování vodíku na platinové destičce z roztoku kyseliny sírové s aktivitou iontů vodíku rovnou jedné.
V řadě napětí jsou výše postavené kovy méně ušlechtilé a nižší kovy ušlechtilejší. Když ponoříme méně ušlechtilý kov do roztoku soli ušlechtilejšího kovu, probíhá při tomto „krátkém chemickém spojení“ ten stejný proces jako v galvanickém článku: kov, který má zápornější potenciál, vytěsní z roztoku kov s kladnějším potenciálem, ale sám přitom přechází do roztoku. Dáme-li například do roztoku síranu měďnatého zinkový nebo železný plíšek, začne se na něm vylučovat měď a zinek nebo železo přechází do roztoku.
Zn(s) + CuSO4(l) → ZnSO4(l) + Cu(s)
Chemický proces, který při tom probíhá, se skládá z odevzdávání elektronů jednoho prvku druhému podle následujících rovnic:
Zn0 + Cu2+ + SO42- → Zn2+ + SO42- + Cu0
Fe0 + Cu2+ + SO42- → Fe2+ + SO42- + Cu0
Podobně je možné z roztoku vytěsnit měď železem, kadmium a zlato zinkem. Tyto způsoby se prakticky používají při srážení kovů z roztoků. Proces založený na reakcích vytěsňování ušlechtilejšího kovu z roztoku méně ušlechtilého se nazývá cementace. V tabulce 9 jsou uvedeny potenciály vybraných kovů.
|
Tabulka 9 : Elektrodové potenciály některých kovů při teplotě 25 ºC vztažené k Pt/H/H+ elektrodě |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Cementace je elektrochemický proces (nazývaný i vnitřní elektrolýza). Termodynamická možnost průběhu cementace je daná velikostí poměru elektrodových potenciálů. Vytěsňující kov musí mít elektrodový potenciál zápornější než vytěsňovaný.
φoMe2 < φoMe1
Protože s postupem cementace se koncentrace kovu v roztoku mění, tím se mění i hodnota potenciálu, bude proces probíhat až do ustanovení rovnováhy potenciálů, kdy
φoMe1 = φoMe1
V některých případech se termodynamické rovnováhy nedosáhne pro kinetické těžkosti. Např. železo se nevycementuje zinkem při normální teplotě (20 ºC), protože při této teplotě je rychlost reakce velmi nízká.
Srážení mědi z roztoku železným šrotem je možné vykonávat periodicky nebo nepřetržitě. Periodické srážení probíhá v bubnech, nádržích a ve válcovitých nádobách – cementačních hruškách. Nepřetržitá cementace se provádí ve žlabech (obrázek 51) nebo cementačních kuželech (obrázek 52).
Cementační žlaby jsou široké od 0,3 do 3 m a hluboké od 0,5 do 1,5 m. Po celé délce žlabu jsou zabudovány přehrádky, aby roztok střídavě protékal pod přehrádkou a nad ní. Na dně žlabu jsou mřížky tvořící pomocné dno na uložení železného šrotu.
|
|
|
Obrázek 51: Cementační žlab |
Cementační měď prochází při natřásání železného šrotu mezerami mřížky, sbírá se na dně žlabu, odkud se periodicky vymývá proudem vody pod tlakem do příčných vynášecích kanálů. Dřevěné cementační žlaby mají možnost regulace úklonu a tím i rychlosti průtoku roztoku a času styku srážedla s roztokem. Plnění i vyprazdňování železného šrotu a cementační usazeniny se obsluhuje ručně.
|
|
|
Obrázek 52 : Cementační kužel |
Cementační kužel je válcovitá nádrž průměru 4,2 m a výšky 7,2 m, ve které je zabudovaný kužel s průměrem a výškou 3 m a obrácený vrcholem dolů. Dno má sklon 45˚, což umožňuje odvod cementační mědi. Horní rozšířená část kuželu je ze síta z nerezavějící ocele, kterým přepadávají odloupnuté částečky mědi a klesají k nakloněnému dnu válce.
Ve spodní části kuželu je přivádějící potrubí rozvětvené na 6 rourek s rýhami tangenciálně nasměrovanými ke stěně kuželu. Tím je umožněno dosáhnout vířivého pohybu při vhánění roztoku s obsahem rozpuštěné mědi. Vnitřní prostor kuželu nad sítem z nerezavějící ocele je vyplněn drceným, odcínovaným železným šrotem. Železo je nahromaděno až nad vrchní okraj válce a jeho velká hmotnost se uplatňuje jako účinný prostředek na udržení teploty, čím se zvyšuje reakční rychlost srážení. Vstřikování výluhu způsobuje nejen rychlé srážení mědi, ale také napomáhá odstraňovat kovovou měď z povrchu železa a tím obnovovat jeho aktivní povrch. Takto získaná cementační měď je čistší než měď z cementačních žlabů. Obsah Cu se pohybuje v rozsahu 90% až 95%, obsahy Fe, Si, Al jsou pod 1 %. Výtěžnost mědi v tomto zařízení dosahuje až 95 %.
Na obrázku 53 je schématicky znázorněno nové kontinuálně pracující vibrační cementační zařízení, které výrobce, firma Klöckner-Humboldt-Wedag, nazval cementační vibrační reaktor. Zařízení připomíná vibrační mlýn. Dvě horizontální válcové nádrže, vzájemně propojené pružným spojovacím hrdlem, jsou pružně uložené na základech a vibrátor je udržuje v kmitavém pohybu.
|
|
|
Obrázek 53: Cementační vibrační reaktor KHW (Německo) |
Do horního válce s částečnou náplní železného šrotu se kontinuální přivádí roztok CuSO4 a železný granulát. Roztok spolu s cementační jemnozrnnou mědí přetéká do spodní válcové nádrže, která je také částečně naplněna šrotem. Ze spodní nádrže vytéká ochuzený roztok a vyplavuje i cementační měď do zásobníku, kde se získává měděný sediment. Reakční rychlost je ve srovnání s jinými cementátory údajně až 100-násobně větší. Výhodami cementačních vibračních reaktorů jsou: nepřetržitý provoz, velká reakční rychlost, poměrně malé investiční a provozní náklady.
K novějším cementačním zařízením patří cementátory s „fluidní vrstvou prášku“ a pulzační cementátory. Fluidní stav vrstvy se vyvolá stoupajícím proudem roztoku přiváděného do spodní části aparatury. Velký specifický povrch kovu cementátoru, velká rychlost přemisťování tuhých částeček, případně kapaliny v důsledku stálé cirkulace částeček a stálé obnovování tuhé fáze podporují intenzifikaci procesu a snižují ztráty prášku.
Vysoce efektivní zařízení je pro cementaci pulzační cementační přístroj, který je zobrazen na obrázku 54. Používá se na cementační očištění zinkového elektrolytu od iontů mědi a kadmia. Roztok, který se má očistit, se přivádí do spodní části kolony, přechází vrstvou granulovaného kovu cementátoru (Zn) a stoupá směrem nahoru k filtru. Vibrací diafragmy se dosahuje velká rychlost toku a současně i odstranění vycementovaného kovu z povrchu granul Zn. Produkty cementace se vynášejí z přístroje spolu s roztokem. Průmyslový pulzační přístroj na očištění zinkového elektrolytu od mědi a kadmia zpracuje při objemu 1 m3 30 m3. h-1 roztoku a sníží obsah mědi z 2 až 6 g.l-1 na stopový obsah.
|
|
|
Obrázek 54: Pulzační cementační přístroj |
Cementace mědi, která se dříve ve značné míře používala, je v současnosti nahrazována, především pro vysokou cenu železného odpadu, jinými metodami. Dosud je důležitým činitelem při srážení ušlechtilých kovů z kyanidových výluhů, při rafinaci roztoků před elektrolýzou, při srážení kovů z velice zředěných důlních vod nebo roztoků po biologickém vyluhování. V poslední době se především při srážení ušlechtilých kovů cementace nahrazuje jejich adsorpcí na aktivní uhlí, hlavně proto, že po provedené desorpci může být aktivní uhlí znovu regenerováno.
V hydrometalurgii neželezných a drahých kovů se využívá krystalizace na vyloučení čistých solí kovů z roztoku, na očištění roztoku od příměsí a případné získání vedlejších produktů.
Termodynamika krystalizace z vodného roztoku, to znamená podmínky rovnováhy krystaly – roztok, se znázorňuje pomocí diagramů rozpustnosti. Při dvousložkových systémech se používají dva typy diagramů rozpustnosti:
a) polytermy rozpustnosti
b) diagramy rozpustnosti.
Polyterma rozpustnosti S je křivka znázorňující závislost rozpustnosti (y –souřadnice) na teplotě (x – souřadnice). Rozpustnost solí ve vodě se vyjadřuje v %, g.l -1 nebo mol.l-1. Pokud je křivka rozpustnosti plynulá, nelomená, nenastávají v systému polymorfní změny. Křivka S obyčejně s teplotou stoupá (obrázek 55 a), někdy se rozpustnost s teplotou mění (obrázek 55 b) a jen zřídka s teplotou klesá (obrázek 55 c). Někdy pozorujeme složitý průběh, který se projeví maximem nebo minimem na křivce.
|
|
|
Obrázek 55: Typy polyterm rozpustnosti |
Když spolu reaguje voda a sůl chemicky, tvoří se krystalohydráty, na křivce – polytermě rozpustnosti se vytvoří zlomy. Příkladem je rozpustnost FeSO4 zobrazená na křivce (obrázek 56). Bod a určuje rovnováhu roztoku s dvěmi druhy krystalohydrátů FeSO4.7H2O a FeSO4.4H2O, bod b pro krystalohydráty FeSO4.4H2O a FeSO4. H2O.
Diagram rozpustnosti zobrazuje závislost složení a teploty. Při tomto typu diagramu se na svislé osy vynáší teplota a na horizontální osy složení roztoku v hmotnostních nebo molárních %, nebo g na 100 g roztoku.
Pro proces krystalizace jsou využitelné dva efekty:
a) zvýšení teploty – odstranění rozpouštědla (odpařením),
b) snížení teploty – snížení rozpustnosti.
|
|
|
Obrázek 56: Polyterma rozpustnosti síranu železnatého |
Proces krystalizace se skládá ze dvou stupňů:
a) tvorba zárodků (krystalizačních středisek),
b) další růst krystalů.
Krystalizační střediska mohou vznikat samovolně (homogenní tvorba krystalizačních středisek), nebo se do roztoku záměrně hodí tuhé látky, které tvoří oporný povrch pro růst krystalů, např. částice Al(OH)3 při vykrystalizování Al(OH)3 z roztoku hlinitanu sodného (heterogenní krystalizace). Ke vzniku velkého počtu krystalizačních středisek přispívá rychlé ochlazení, intenzivní míchání, přítomnost ostrých bodů (povrchů), velká čistota roztoků a velká koncentrace.
Při velkém počtu krystalizačních středisek se každé z nich nedostatečně nasycuje rozpustnou látkou, a tak vznikají drobné krystalky. Když probíhá krystalizace při pomalém ochlazování, počet krystalizačních středisek je malý a vznikají velké krystaly.
Ve většině případů probíhá krystalizace pozvolna a ke konci procesu je koncentrace mateřského louhu stejná jako nasyceného roztoku při konečné teplotě krystalizace.
Jsou-li v roztoku současně dvě nebo více látek, může se proces krystalizace řadit tak, že krystalizuje jen jedna z nich, ostatní zůstávají v roztoku. Tyto metody frakční krystalizace jsou založeny na různé rozpustnosti solí.
Využití krystalizace ochlazováním je výhodné, když rozpustnost látky v kapalině rychle klesá se snížením teploty. Krystalizace ochlazováním se používá dost často, protože vypařování roztoku bez varu probíhá pozvolna a je drahé. Postup krystalizace z roztoku o koncentraci co snížením teploty z To na T2 je možné sledovat na obrázku 57. Teplota To a koncentrace rozpuštěné látky v roztoku c jsou souřadnicemi bodu 1 nad křivkou rozpustnosti. Bod 1 se nachází v oblasti dobré rozpustnosti. Při snižování teploty z hodnoty To při koncentraci co na hodnotu T dostaneme křivku rozpustnosti v bodě 2, kde začíná tvorba krystalů.
|
|
|
Obrázek 57: Postup krystalizace ochlazováním roztoku |
Dalším snižováním teploty z hodnoty T1 na T2 přicházíme na křivce rozpustnosti do bodu 3, kterému odpovídá už podstatně nižší rozpustnost tuhé fáze, charakterizovaná nižší kritickou rozpustností c1. Podíl tuhé látky, odpovídající rozdílu koncentrací co – c1, se vyloučil z roztoku ve formě krystalů.
Krystalizace odstraněním rozpouštědla: Rozpouštědlo se může odstranit buď při teplotách nižších než je teplota varu, nebo za varu. Intenzívnost odstranění rozpouštědla ovlivní vznikající počet krystalizačních středisek, rychlost růstu a velikost krystalů.
V provozu se uskutečňuje krystalizace v pánvích – krystalizátorech, s rozsáhlým povrchem hladiny vypařování, protože při tom vzrůstá množství vypařující se kapaliny. Pánvové krystalizátory jsou přístroje s periodickým provozem. Plní a čistí se ručně nebo mechanicky. Při volném vypařování (bez přívodu tepla) se roztok na povrchu taky ochlazuje a v souvislosti s tím dodatečně zvyšuje stupeň přesycenosti roztoku. Rozpouštědlo se může rychleji odstraňovat odpařováním přiváděním tepla, to se v praxi využívá častěji.
Krystalizace ochlazováním: Tento způsob vlastně spolupůsobí při krystalizaci, při které se rozpouštědlo odstraňuje volným způsobem. Tady se roztok ochlazuje odebráním výparného tepla, které je v něm skryté. Pokud se současně nepřivádí teplo, probíhá odpařování na úkor odebrání tepla kapalině.
Při zpracování rud neželezných kovů hydrometalurgickým způsobem se krystalizace používá velmi zřídka. V současnosti se krystalizace používá při loužení manganových rud oxidem siřičitým na krystalizaci síranu manganatého. Rozdílná rozpustnost složek se využívá při čištění roztoků frakční krystalizací.
Hydrolýza se v chemické úpravě rud velmi rozšířila. Používá se především při srážení hydroxidů z roztoku a často se využívá při čištění roztoků před elektrolytickým srážením. Ve formě hydroxidů se sráží z roztoku např. kobalt a některé jiné kovy.
Úpravou hodnoty vodíkového exponentu (pH) je možné z roztoku selektivně vysrážet různé kovy ve formě hydroxidů. Tak např. při zpracování roztoků získaných při loužení pyritových výpalků po chloridovém pražení se vykonává frakční srážení hydroxidu železa při pH = 4 až 4,5, hydroxidu kobaltu při pH = 5 až 5,5 a hydroxidu zinku pH = 6 až 8
MpH = 4 až 4,5
FeCl3 + 3 H2O → Fe(OH)3 ↓ + 3HCl
MpH = 5 až 5,5
CoCl2 + 2 H2O → Co(OH)2 + 2 HCl
pH = 6 až 8
ZnCl2 + 2H2O → Zn(OH)2 + 2 HCl
Podle Rotiniana a Chejfica se hydroxidy kovů vysrážejí z roztoků při určitém pH, jehož hodnota je funkcí aktivity L náboje kovu v roztoku (tabulka 10).
|
Tabulka10: Parametry srážení hydroxidů některých kovů z roztoků |
||||||||||||||||||||||||||||||||||||
|
Pokud využíváme hodnoty pH při frakčním srážení kovů ve formě hydroxidů, musíme brát v úvahu, že pH závisí na koncentraci daného kovu v roztoku a také na typu aniontů v roztoku.
Dalším ze způsobů získávání kovů z roztoků je srážení chemickými činidly. Jako příklad je možno uvést sulfidové srážení kovů, kdy jsou kovy převáděny do formy nerozpustných sulfidů.
Me2+ + Na2S → MeS + 2 Na+
K nejpoužívanějším činidlům patří sulfid sodný (Na2S), železnatý (FeS) a vápenatý (CaS). Předností sulfidového srážení je velmi nízká rozpustnost sulfidů kovů, díky které je možné dosáhnout vysoké účinnosti odstranění kovů se zbytkovými koncentracemi např. pro Cd nebo Zn (0,01 mg/l).
Další z možností odstraňování kovů z vod je uhličitanové srážení. Tento proces je zvlášť výhodný pro některé dvojmocné kovy (jako je například kadmium), které tvoří málo rozpustné uhličitany již při nižší hodnotě pH, než je potřebná pro srážení hydroxidové. Kal vzniklý při uhličitanovém srážení je hutnější, než kal po hydroxidovém srážení a má lepší sedimentační a filtrační vlastnosti. K výhodám procesu je možno přičíst i schopnost udržet nízké zbytkové koncentrace kovů v poměrně širokém rozmezí pH, což svědčí o malé citlivosti procesu na změny v dávkování uhličitanu sodného.
Srážecí proces kovových oxidů v destilátorech se v hydrometalurgii používá při zpracování roztoků, které obsahují komplexní uhličitany mědi a niklu.
Nejrozšířenější je metoda srážení oxidu měďnatého v destilátorech z roztoků, získaných při loužení měděných rud (oxidovaných i obsahujících ryzí měď) čpavkovými roztoky.
Destilací roztoků, které obsahují komplexní amoměďnatou sůl, se získá plynný NH3 a CO2 a sraženina černého oxidu měďnatého. Amoniak i oxid uhličitý se zachycují a používají se znovu jako činidlo při loužení rud. Sraženina mědi se dopravuje do závodů na přetavování mědi na další zpracování. Loužení uhličitanem amonným a čpavkem.
CuCO3. Cu(OH)2 + (NH4)2CO3 + 6 NH3 → 2 Cu(NH3)4CO3 + 2 H2O
Cu(NH3)4CO3 → CuO + 4 NH3 + CO2
Komplexní uhličitan měďnatoamonný se destilací rozkládá už pod 100 ºC.
Adsorpci je možno charakterizovat jako zachycování rozpuštěných nebo koloidních látek na povrchu tuhé fáze. V praxi se využívá například k odstraňování zbytkového znečištění z vody nebo k zachycování biologicky nerozložitelných či toxických látek. Mezi nejčastěji používané sorbenty patří hydratované oxidy železa nebo hliníku, aktivní uhlí, koks, škvára, popílky, hlinitokřemičitany, rašelina a jiné. Jako sorbentu je možné použít i vyřazených automobilových pneumatik. Bylo zjištěno, že s jejich pomocí lze odstranit většinu kovů z vodných roztoků stejně efektivně jako při použití aktivního uhlí.
Adsorpci rozpuštěných kovů ovlivňuje mnoho faktorů. Patří k nim chemická forma kovu (oxidační stav, stupeň komplexace), pH roztoku, přítomnost cizích iontů, teplota a charakter adsorbentu. Rozhodující význam při adsorpci má však velikost a jakost povrchu adsorbentu, který nese tzv. aktivní centra.
Jak už bylo řečeno, v České republice se v současnosti získávají ušlechtilé kovy pouze dovozem nebo zpracováním druhotných surovin. Zásoby některých nerostných surovin, vyskytujících se na našem území, byly do značné míry vyčerpány a těžba tudíž ukončena. Jedinou rudou, která se v současnosti u nás dosud těží, je uran.
Klasické metody zpracování druhotných surovin s obsahem ušlechtilých kovů s využitím pyrometalurgických technologií při výrobě olova a následné zpracování a rafinace získaných meziproduktů, ve kterých se ušlechtilé kovy koncentrují, jsou detailně uvedeny v celé řadě publikací zaměřené na výrobu neželezných kovů.
Odpady s obsahem zlata a stříbra představují širokou škálu různých typů a forem odpadů s velmi proměnlivým zastoupením ušlechtilých kovů, proto je jejich zpracování možné pouze opět rozmanitou řadou technologií a kombinovaných metod. Samotná volba vhodné technologie úpravy odpadů je dána jejich kovnatostí, obsahem doprovodných prvků a složením ostatního průvodního materiálu. Je proto nutnost vždy posuzovat možnost komplexního využití všech složek odpadů.
Při vhodné volbě recyklační technologie se vždy vychází ze schémat pro zpracování druhotné suroviny s předběžnou mechanickou a tepelnou úpravou s následným pyrometalurgickým, hydrometalurgickým nebo nejčastěji kombinovaným zpracováním (obrázek 58).
|
|
|
Obrázek 58: Zjednodušené schéma zpracování primárních a sekundárních surovin |
Proces recyklace odpadů s obsahem ušlechtilých kovů je ve srovnání s možnostmi zpětného získávání ostatních neželezných kovů nepoměrně těžší, neboť pro největší podíl odpadů platí, že:
· Obsah ušlechtilých kovů tvoří stále nižší podíl výrobků (trvalá tendence)
· Výrobky mají vzhledem k miniaturizaci stále komplikovanější složení
Klasické způsoby recyklace již neodpovídají potřebám a je nutno je zdokonalovat a rozšiřovat o nově vypracované metody.
Struktura elektroodpadu se neustále mění. Stále více sílí požadavek na zdokonalení a účinnou stimulaci sběrové cesty a zejména na důsledné dělení jednotlivých frakcí. Přepracování a likvidace elektroodpadu představuje velmi rozsáhlý soubor činností aplikovaných na neobyčejně rozmanitou třídu materiálů, která zahrnuje předměty od velikosti mobilního telefonu až po mnohatunové průmyslové soubory jako velíny se souvisejícími systémy čidel, telefonní ústředny nebo velkokapacitní chladící zařízení (obrázek 59).
|
|
|
Obrázek 59: Ukázka elektroodpadu |
Jednotlivé komponenty těchto zařízení obsahují širokou škálu použitých materiálů. Jedná se o plasty, sklo, ale hlavně neobyčejně pestrou směs kovů a polovodičových materiálů od těch nejběžnějších až po zlato,stříbro, platinu anebo takové prvky jako Ta, Co, Nd, Eu nebo Sm. Není snad nutno říkat, že tyto odpady jsou svým složením zátěží pro životní prostředí (obsah těžkých kovů, elektrolytů, freonů apod.) a patří tudíž do kategorie nebezpečný odpad. Elektroodpad však také obsahuje cenné materiály, jejichž opětovné využití může proces recyklace učinit ekonomicky soběstačným nebo dokonce významně ziskovým. Velkým problémem recyklačních technologií při získávání obecných a barevných kovů (Fe, Cu, Pb, Al, atd.) je to, že jednotlivé součástky elektrotechnických zařízení s obsahem kovů jsou obyčejně drobné a s mnoha propojeními, jejich segregace je tedy náročná.
Jednotlivé metody zpracování elektroodpadu lze rozdělit takto:
Mechanické metody
Pyrometalurgické metody
Hydrometalurgické metody
Elektrochemické metody
Biotechnologické metody
Principem mechanických metod recyklace ušlechtilých kovů je zmenšení velikosti vstupního materiálu a jeho roztřídění s využitím fyzikálních vlastností jednotlivých složek. První operací, která předchází mechanickému zpracování, bývá u elektroodpadu většinou ruční nebo částečně mechanizovaná demontáž zaměřená na součásti s obsahem cenných kovů jako jsou transformátory, cívky, chladící tělesa, motory, kondenzátory, baterie, kabely, vodivé desky apod. Rovněž musí být ručně odstraněny součásti s obsahem nebezpečných látek, například rtuťové spínače, baterie, kondenzátory s obsahem PCB a jiné.
Pyrometalurgické metody využívají procesů pyrolýzy, tavení, spékání (aglomerace) a reakce s plynnou fází za vysokých teplot. Klasické způsoby zpětného získávání ušlechtilých kovů pyrometalurgickými metodami se zásadně neodlišují od procesů používaných při zpracování meziproduktů s obsahem ušlechtilých kovů z prvovýroby neželezných kovů z koncentrátů. Pokud obsahuje materiál plastické hmoty nebo jiné organické složky, pak nejběžnějším způsobem jejich odstraní je spálení v tavenině kovů nebo v peci.
Elektrotechnické součástky jako jsou konektory, tištěné spoje nebo integrované obvody se mísí v peci s roztaveným olovem. Plasty vyhoří, železo a část barevných kovů plavou na povrchu taveniny a odtud se stahují. Do roztaveného olova přechází většina ušlechtilých kovů. Tavenina se následně prohání vzduchem, většina olova a obecných kovů se zoxiduje a odstraní jako struska. Zbylá část olova obohacená o drahé kovy se podrobí rafinaci. Hlavní výhodou tavících procesů je schopnost zpracovat všechny formy elektronického odpadu,
nevýhodou pak nepříliš dobrá ekologická šetrnost – odplyny z hoření plastů, struska s obsahem těžkých kovů atd.
Dalším řešením recyklace elektroodpadu je termické spálení v pyrolýzním bubnu (rotační peci). Šnekovým podavačem se rozmělněné komponenty dávkují do pece spolu s koksem. Čištění plynu se provádí ve Venturiho pračkách pomocí s přídavkem vápenného mléka. Pevné produkty procesu jsou složeny ze směsi železa, neželezných kovů, skelných vláken s koksem a keramiky.
Další z metod je reakce s plynnou fází, která se využívá při zpracování drcených zlacených elektronických součástek. Jedná se o chloraci plynným chlórem při teplotě 500˚C a době chlorace 20 minut. Výsledným produktem je AuCl3. Byla testována i metoda přímé chlorace a bromace na postříbřených kontaktech z mosazi.
Hlavním technologickým uzlem hydrometalurgických procesů druhotných surovin bývá většinou několikastupňové kyselé, zásadité nebo amoniakální loužení už roztříděných materiálů. Získané výluhy jsou zpracovány na kov, respektive chemický koncentrát celou řadou procesů počínaje cementací, precipitací, destilačním srážením až po iontovou výměnu, extrakci a extrakční elektrolýzu. Samotná elektrolýza je pro složitost odpadních roztoků, které obsahují velké množství ekonomicky i ekologicky náročně zpracovatelných kovů ( Cu, Zn, Ni, Cd, Ag, Pd, Fe, atd. ) používána ve světě pro přepracovávání elektroodpadu poměrně zřídka. Většinou se elektrolýzou získává podíl mědi případně niklu a drahé kovy zůstávají v anodických kalech.
Mezi výhody hydrometalurgických metod při zpracování druhotných surovin patří:
· Možnost zpracovávat chudé i bohaté materiály (obsahy kovů od g/t až do 10kg/t)
· Procesy probíhají při nízkých teplotách
· Dosažení vyšší výtěžnosti a menších ztrát ušlechtilých kovů, než u pyrometalurgických metod
· Nižší provozní náklady a spotřeba energie
· Lepší možnost ochrany životního prostředí
Mezi nevýhody patří:
· Nízká životnost zařízení vzhledem k agresivitě používaných chemikálií
· Používání drahých konstrukčních materiálů (antikorozní ocel, titan, sklo)
· Vznik silně zasolených odpadních vod s obsahem Cl-, NO3-, SO42-
· Exhalace s obsahem NOx, SO2
· Neschopnost okamžitě reagovat na změnu složení zpracovávaného materiálu
· Velké objemy loužících roztoků
Spojovací články, konektory, tištěné spoje a jiné komponenty jsou zpracovávány dle následujícího postupu. Nejprve je převeden měděný nosič s povlakem ušlechtilých kovů do roztoku kyselým loužením. Jako loužící činidlo se používá vodný roztok H2 SO4 a CuSO4. Uvolněné povlaky ušlechtilých kovů jsou odseparovány filtrací. Výluh je zpracován na kovovou měď cementací. Takto získaná měď je následně přetavena a přečištěna elektrolytickou rafinací. Získanými produkty jsou ušlechtilé kovy v uvolněných povlacích, anodové kaly s obsahem 12,81 g/kg stříbra, 0,036 g/kg zlata a katodová měď čistoty 3N.
Nejvíce zastoupený a také nejžádanější kov přítomný v elektroodpadech – zlato, je možno selektivně a snadno izolovat loužením zředěnými (0,1%) roztoky alkalických kyanidů, především NaCN. Podmínkou je, aby byl pozlacený materiál obnažen a jeho povrch byl přístupný kontaktu s loužícím roztokem. Nezbytná je také přítomnost vzdušného kyslíku.
4 Au + 8 NaCN + 2 H2O + 02 → 4 NaAu(CN)2 + 4 NaOH
Z kyanidového roztoku se zlato sráží dnes hlavně zinkovými hoblinami nebo prachem, podobně jako u stříbra.
2 NaAu(CN)2 + 2 Zn + 4 NaCN + H2O → 2 Na2Zn(CN)4 + 2 Au + 2 NaOH + H2
Loužení má vysokou účinnost a jeho výhodou je, že ostatní kovy nejsou kyanidy rozpouštěny. Nejčastěji používané slitiny na bázi Cu, Zn, Ni tak mohou být dále metalurgicky rafinovány, aniž by tyto kovy přecházely do roztoků a z nich musely být složitě izolovány.
Nevýhodou je samozřejmě vysoká toxicita použitého činidla. Při správném chemickém zacházení s výluhy je však toxických odpadů minimální množství a dají se jednoduše rozložit na uhličitanové a amonné ionty, například oxidací chlornanem:
CN- + ClO- → CNO- + Cl-
K detoxikaci méně toxických kyanátových iontů lze pak použít aeraci nebo reakci s chlornanem:
2 CNO- + 3 ClO- + H2O → 2 CO2 + N2 + 3 Cl- + 2 OH-
Provozní rizika a potencionální možnost havárie však tento proces činí problematickým.
Dalším ze způsobů loužení zlata spočívá v převodu Au do roztoku ve formě komplexu Au[SC(NH2)2]2+ loužením směsným činidlem. Optimální složení loužícího činidla je 15g/dm3 thiomočovina, 10% H2SO4 a 5g/dm3 Fe2(SO4)3.
2Au + Fe2(SO4)3 + 4SC(NH2)2 → {Au[SC(NH2)2 ]2}SO4 + 2FeSO4
Kromě zlata a stříbra se v elektroodpadu vyskytuje mnoho dalších ušlechtilých kovů. Paladium se v elektroodpadech vyskytuje ve třech hlavních aplikacích – v nejiskřících kontaktech ( relé, stykače ), jako náhražka zlata na povrchu mechanických kontaktů anebo v deskových keramických kondenzátorech. Vzhledem k značné chemické příbuznosti k mědi je jeho elektrochemická izolace málo účinná, srážení některých málo rozpustných komplexních sloučenin paladia rovněž nedává uspokojivé výsledky. Pokud se ale surovina s obsahem paladia zpracovává sulfáto – nitrátovou cestou, vzniklé roztoky lze po denitraci velmi snadno redukovat formaldehydem a získat s vysokou účinností paladium. Procesu vadí přítomnost halogenidových iontů.
V harddiscích počítačů, ale i např. v reproduktorech audiozařízení se stále více nacházejí místo klasických magnetů nebo feritů kompozitní materiály s velmi vysokou magnetickou susceptibilitou na bázi Sm, Co, B nebo Nd-Fe-B. Izolace těchto elementů není ani tak ekonomickým přínosem, ale hlavně technologickým požadavkem. Obsah lanthanoidů silně zhoršuje kvalitu železné složky šrotu a navíc způsobuje potencionální riziko vzhledem k pyroforičnosti a jiskřivosti těchto slitin. Při kvalitní mechanické demontáži mohou být tyto komponenty snadno odstraněny a navíc poměrně jednoduchým chemickým procesem mohou být cenné složky recyklovány.
Recyklace televizních obrazovek a PC monitorů je objemově dosti významnou součástí procesu přepracování elektroodpadů. Specifická je přitom právě přítomnost skleněné obrazovky. Sklo je masivní z důvodů podtlaku v obrazovce, takže tvoří 1/3 až 1/2 hmotnosti celého přístroje. Stínítko obrazovky je vyráběno z barnatého nebo strontnatého skla, kónus z olovnatého skla. Pro opětovné využití skla je nezbytné zbavit obrazovky luminiscenční vrstvy nanesené na vnitřní straně. Luminofor je jednak toxickým odpadem pro obsah těžkých kovů a S, ale navíc znemožňuje opětovné využití skla tím, že významně mění optické vlastnosti skla. Obrazovkové sklo je tedy čištěno mokrou nebo suchou cestou, při obou vzniká kal či prach luminoforu. Ten je dosud deponován jako toxický odpad bez přepracování, přestože obsahuje yttrium (10%) a europium (2-3%), jejichž cena se pohybuje v řádu tisíců , respektive desetitisíců Kč/kg. V současnosti probíhá poloprovozní ověřování metody recyklace těchto kovů, čímž by se ČR zařadila mezi nejpokročilejší státy v této oblasti.
Závěrem lze konstatovat, že recyklace elektroodpadu představuje perspektivní obor, o který v budoucnu, vzhledem k rostoucí ceně kovů na světových trzích, zcela zřejmě poroste zájem. Základní pyrometalurgické a hydrometalurgické technologické postupy budou sloužit ke zpracování elektroodpadů a zároveň budou muset splňovat přísná ekologická měřítka.
Galvanické povrchové úpravy jsou komplexním zdrojem plynných, kapalných a tuhých škodlivin. Velká pozornost je věnována problematice odpadních oplachových vod z hlediska vzniku kapalných škodlivin, spotřeby vody a snížení ztrát cenných chemikálií. Ztráty elektrolytu z galvanických lázní do oplachových vod jsou 100 - 400 ml z 1 m2 pokovené plochy. Množství ztrát vzniklých při pokovování ukazuje tabulka 11.
|
Tabulka 11: Roční hmotová bilance vypočtená pro 38 000 m2 pokovené plochy |
||||||||||||||||||||||||
|
Galvanické kaly obsahují celou řadu škodlivin jako jsou:
Kyanidy
Kyseliny
Odmašťovací soli
Soli nikelnaté
Soli měďnaté
Soli zinečnaté
Soli kademnaté
Soli chromové
Ve stávajících galvanických provozech převládají při detoxikaci odpadních vod dosud klasické neutralizační stanice, kde při neutralizaci vznikají odpadní galvanické kaly. Tyto kaly obsahují zpravidla hydroxidy těžkých kovů (Cu, Ni, Zn, Cd, Cr, Fe) a nerozpustné sloučeniny vápníku ve formě uhličitanů nebo fosforečnanů. Galvanické kaly jsou tak druhotnou surovinou s obsahem kovů potřebných pro národní hospodářství.
Vzhledem k nezanedbatelnému množství kovů, které se nenávratně ztrácejí v galvanických kalech a vzhledem k jejich nepříznivému ekologickému působení byly a jsou neustále prověřovány postupy jejich zpracování. Při všech těchto postupech je sledována jednak selektivní rekuperace obsažených kovů, jednak převod kalů na substrát ekologicky nezávadný či na chemický koncentrát ekonomicky využitelný.
Všem způsobům zpracování je společné zásadité nebo kyselé loužení kalu za účelem převedení těžkých kovů do výluhu. Většina balastních látek se v daném prostředí buď nerozpouští nebo se převádí do formy obtížně rozpustných sloučenin. Získaný výluh se zpracovává selektivními hydrometalurgickými metodami za účelem separace obsažených kovů.
· Cementací (Cu)
· Elektrolýzou (Cu)
· Extrakcí (Cu, Zn)
· Srážením fosforečnany (Fe, Cr, Ni)
· Amoniakální uhličitanové loužení (Cu, Ni, Zn)
· Zkoncentrování výluhů na ionexech (Cu, Zn, Cr, Ni, Cd)
Cementace
Měď se získává z výluhu cementací granulovaným zinkem při pH 1 podle reakce:
Zn0 + Cu2+ → Zn2+ + Cu0
Elektrolýza
Výluh je podroben elektrolýze za podmínek U= 2,5 V, I= 0,07 A. Za těchto podmínek lze získávat měď s 50 – 60 %-ní výtěžností.
Extrakce
Produkty reextrakce jsou pak zinkový koncentrát, niklový koncentrát a měděný koncentrát. Získané koncentráty separovaných kovů se pak poměrně snadno zpracovávají elektrolýzou nebo krystalizací na síranové soli (skalice).
Srážení fosforečnany
Tato metoda je založena na skutečnosti vyplývající z diagramu rozpustnosti, že fosforečnany kationtů s oxidačním stupněm 3+ (Fe3+, Cr3+) jsou v kyselém prostředí méně rozpustné, než fosforečnany kationtů s oxidačním číslem 2+. Morfologie sraženin fosforečnanů na rozdíl od hydroxidů umožňuje snadnou separaci tuhé fáze od kapalné s minimální kontaminací povrchu sraženin jinými rozpuštěnými částicemi.
Amoniakální uhličitanové loužení
Po loužení přecházejí kovy jako jsou Cu, Ni a Zn do výluhu ve formě aminokomplexů [(MeNH3)4]2+. Měď a nikl jsou z výluhu separovány selektivní extrakcí, zinek je pak vysrážen jako uhličitan.
Zkoncentrování výluhů na ionexech
Po kyselém loužení galvanického kalu, který obsahuje kovy vázané ve formě hydroxidů, je výluh podroben sorpci na silně kyselém ionexu. Při průchodu roztoku matricí dochází k sorpci všech účelových kovů na ionex. Při eluci 10%-ní H2SO4 pak kovy přechází do eluátu.
Me(OH)2 + H2SO4(5%) → MeSO4 + 2 H2O (kyselé loužení)
1. Krištofová, D.: Recyklace ušlechtilých kovů. VŠB-Technická univerzita Ostrava, Ediční středisko VŠB-TUO, Ostrava, 2001, ISBN 80-7078-939-5
2. Palas, M.: Nerostné suroviny první poloviny 21. století- současný stav, problémy a perspektivy. Mineral Raw Materials and Mining Activity of the 21st Century. VŠB-Technická univerzita Ostrava, 2003, ISBN: 80-248-0492-1
3. Špaldon, F.: Úprava nerastných surovín, Alfa, Bratislava 1983
4. Jackson, E.: Hydrometallurgical Extraction and Reclamation. Department of Metals and Materials Engineering Sheffield City Polytechnic, 1986.
5. Kušnierová, M.: Biohydrometalurgia. Banícky ústav SAV, Košice, 1992
6. Cvetnaja metallurgija nakanune XXI veka: Sborník vědeckých prací „Gincvetmet“, Moskva 1998
7. Štofko, M., Štofková, M.: Neželezné kovy. Monografie, Košice 2000. ISBN 80-7099-527-0
8. Jursík, F.: Anorganická chemie kovů. Skriptum, VŠCHT Praha, 2002, ISBN 80-7080-504-8
9. Beránek, M., Šebková, J., Pedlík, M.: Technologie kovových materiálů, Praha, SNTL, 1984. 333s.
10. Moore, J.J.: Chemical Metallurgy, (2nd Ed.)
11. Habashi, F.: Textbook of Hydrometallurgy, (2nd Ed.)
12. Hydrometallurgy: A Colection of Invited Papers Dedicated to Professor Nikola Pacovic in Honour of his 65th Birthday. Bor, Yugoslavia, 1996
13. Pacovic, N.: Hydrometalurgija. Radna organizacija Štampa, radio i film, Bor, Yugoslavija, 1980
14. Kraus, S.: Metale pretioase-vol I. Matrix Rom, Bucuresti, Romania, 2006, ISBN 973-755-018-8
15. Kraus, S.: Metale pretioase-vol II. Matrix Rom, Bucuresti, Romania, 2006, ISBN 973-755-019-6
16. Polkin, S., I., Najfonov, T.,V.,: International Mineral Processing Congress, Vol 1, 1965
17. Polkin S., I., Adamov, E., V., Panin, V., V.: Technologia bakteriałnogo vyscelascivanija cvetnych i redkych metallov. Medra, Moskva 1983
18. Fečko, P., Kušnierová, M., Čablík, V., Pečtová, I.: Environmental biotechnology. VŠB-Technická univerzita Ostrava, Ediční středisko VŠB-TUO, Ostrava 2006, ISBN 80-248-1090-5
19. Kušnierová, M., Fečko, P.: Minerálne biotechnologie I. VŠB-Technická univerzita Ostrava, Ediční středisko VŠB-TUO, Ostrava 2001, ISBN 80-248-0023-3
20. Luptáková, A., Kušnierová, M., Fečko, P.: Minerálne biotechnologie II. VŠB-Technická univerzita Ostrava, Ediční středisko VŠB-TUO, Ostrava 2002, ISBN 80-248-0114-0
21. Fečko, P., Kušnierová, M., Sochorková, A., Praščáková, M., Ovčaří, P., Mucha, N., Janáková, I.: Biotechnologie v úprave uhlia. VŠB-Technická univerzita Ostrava, Ediční středisko VŠB-TUO, Ostrava 2008, ISBN 978-80-248-1701-9
22. Habashi, F.: Handbook of extractive metallurgy. Vol. 1, The metal industry, ferrous metals. Wiley-VCH, Weinheim 1997, ISBN 3-527-28792-2
23. Habashi, F.: Handbook of extractive metallurgy. Vol. 2, The metal industry, ferrous metals. Wiley-VCH, Weinheim 1997, ISBN 3-527-28792-2
24. Habashi, F.: Handbook of extractive metallurgy. Vol. 3, The metal industry, ferrous metals. Wiley-VCH, Weinheim 1997, ISBN 3-527-28792-2
25. Habashi, F.: Handbook of extractive metallurgy. Vol. 4, The metal industry, ferrous metals. Wiley-VCH, Weinheim 1997, ISBN 3-527-28792-2
26. Atkins, P.: Periodické království – cesta do země chemických prvků. Akademie věd ČR, Academia, Praha 2005, ISBN 80-200-1185-4
www:
http://old.lf3.cuni.cz (Ústav biochemie, buněčné a molekulární biologie UK Praha)











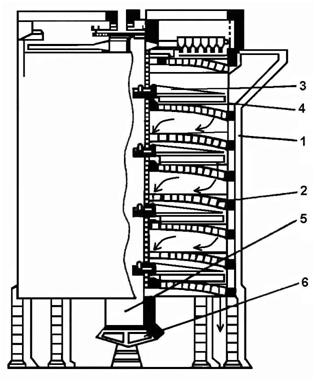
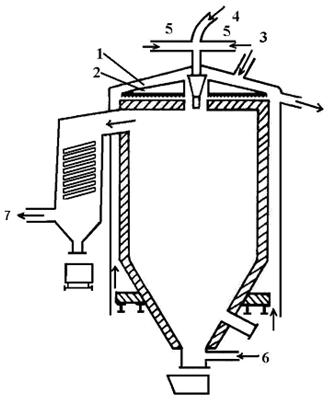
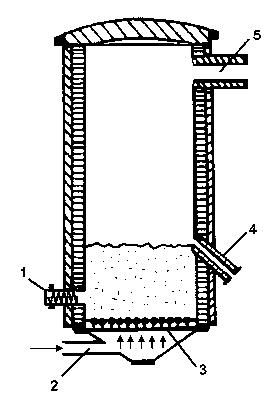

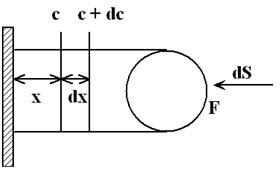
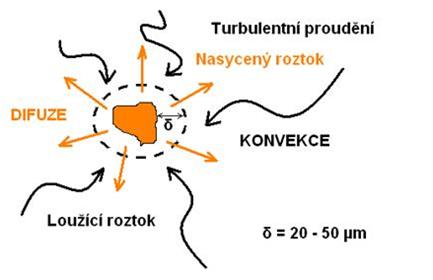
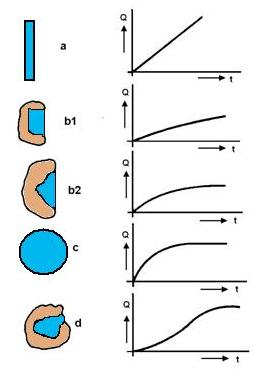
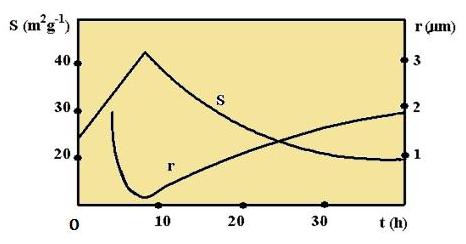
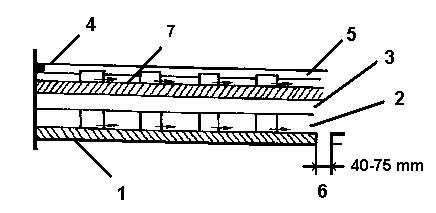
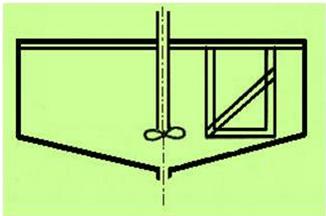
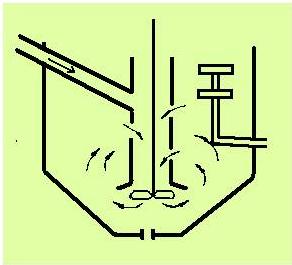
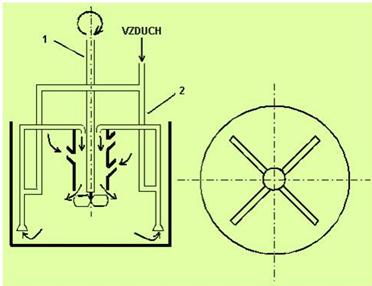

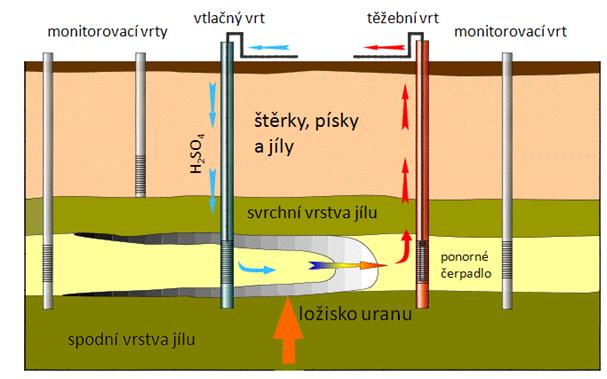
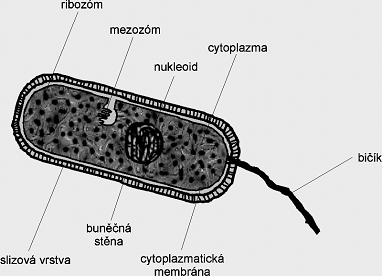
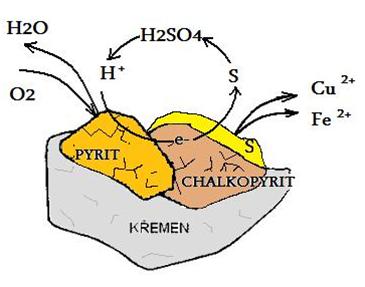
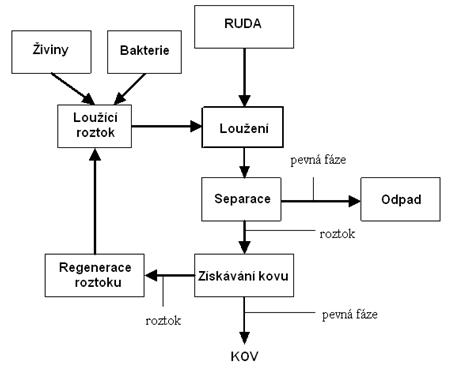
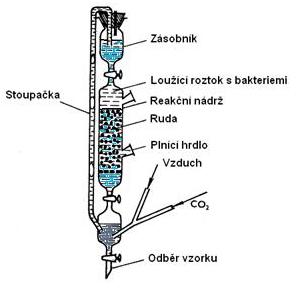
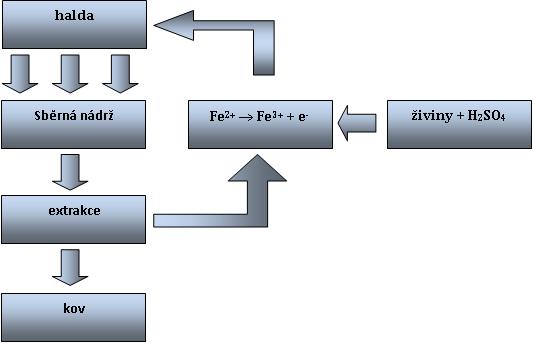
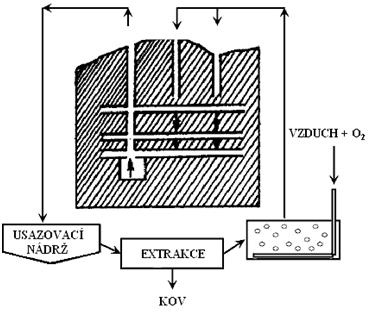


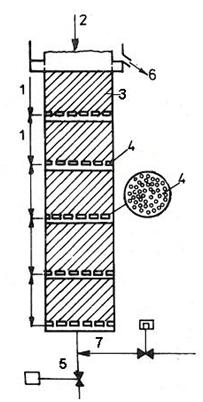
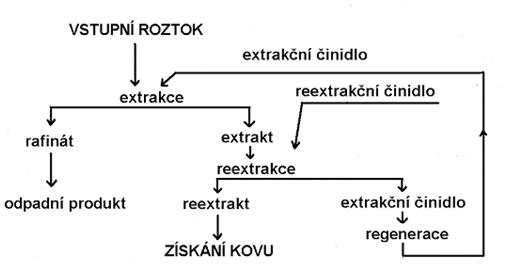
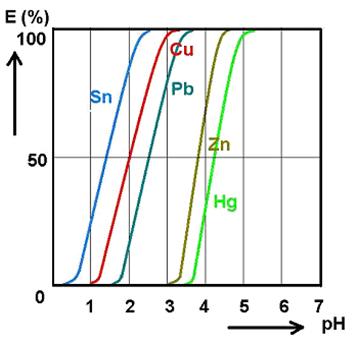
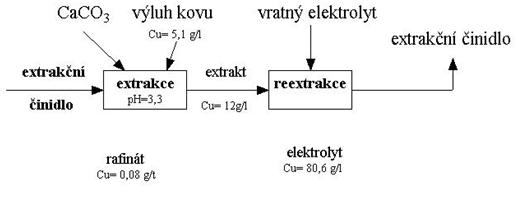
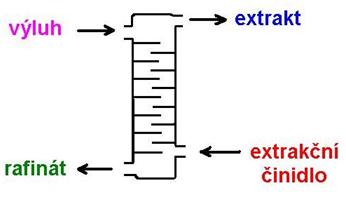
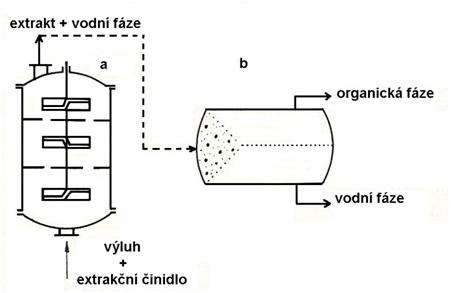
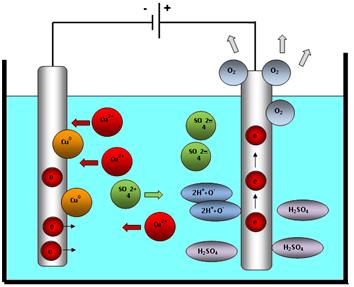
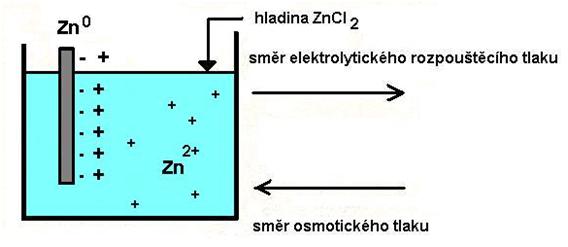
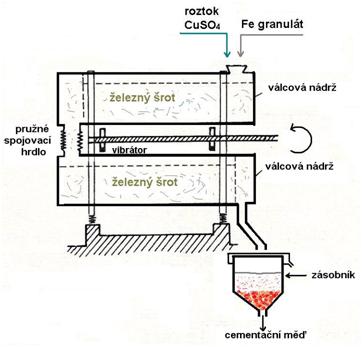
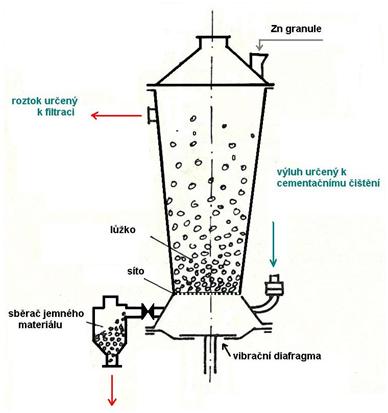
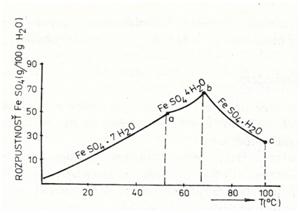
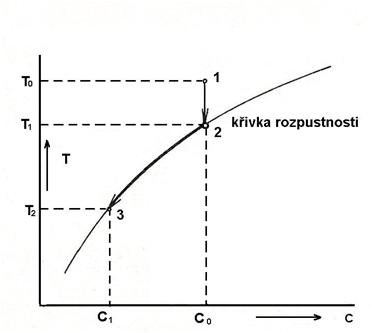
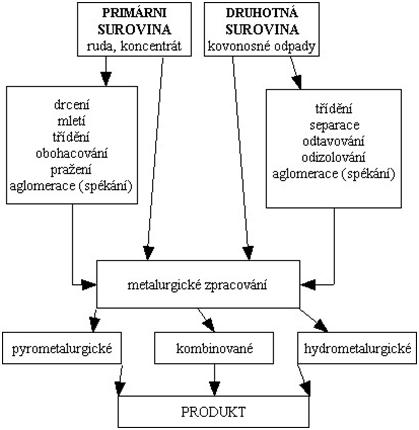

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}